动动发财的小手,点个赞吧!
1. 寻找差异区域
然而,识别特定于细胞系或条件的峰并不能捕获表观遗传事件的全部变化。
为了识别表观遗传事件的差异,我们可以尝试量化 IP 样本中非冗余峰组中片段丰度的变化。
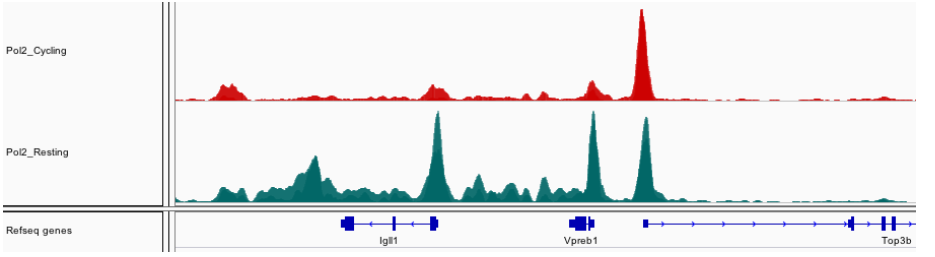
我们首先必须建立一组区域,在这些区域中量化 IP ed 片段。
一种成熟的技术是产生一组非冗余峰,这些峰出现在至少一个被评估的实验条件的大多数中。
在这里,我们确定了在 Mel 或 Ch12 细胞系的两个重复中出现的峰。
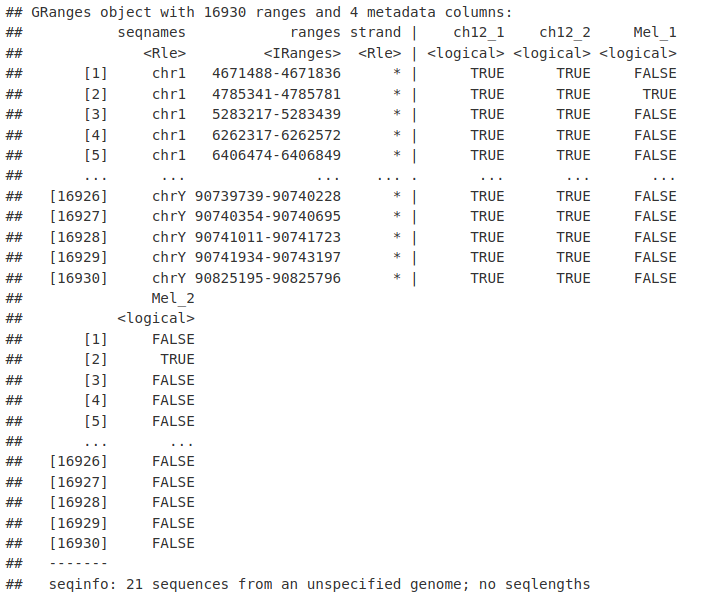
HC_Peaks <- allPeaksSet_nR[rowSums(as.data.frame(mcols(allPeaksSet_nR)[, c("ch12_1",
"ch12_2")])) >= 2 | rowSums(as.data.frame(mcols(allPeaksSet_nR)[, c("Mel_1",
"Mel_2")])) >= 2]
HC_Peaks
export.bed(HC_Peaks, "HC_Peaks.bed")
2. 计数区域
我们将从对齐的 BAM 文件中计数以量化 IP 片段。
正如我们之前所见,我们可以使用 BamFileList() 函数来指定要计数的 BAM,重要的是,为了控制内存,我们使用 yield() 参数指定一次要保存在内存中的读取次数。
library(Rsamtools)
bams <- c("~/Projects/Results/chipseq/testRun/BAMs/Sorted_Myc_Ch12_1.bam", "~/Projects/Results/chipseq/testRun/BAMs/Sorted_Myc_Ch12_2.bam",
"~/Projects/Results/chipseq/testRun/BAMs/Sorted_Myc_Mel_1.bam", "~/Projects/Results/chipseq/testRun/BAMs/Sorted_Myc_Mel_2.bam")
bamFL <- BamFileList(bams, yieldSize = 5e+06)
bamFL
我们可以使用 summarizeOverlaps 函数计算与峰重叠的片段数。由于 ChIPseq 是无链的,我们将 ignore.strand 参数设置为 TRUE。
返回的对象是一个熟悉的 RangedSummarizedExperiment,其中包含我们的非冗余峰的 GRanges 以及我们 BAM 文件在这些区域中的计数。
library(GenomicAlignments)
myMycCounts <- summarizeOverlaps(HC_Peaks, reads = bamFL, ignore.strand = TRUE)
class(myMycCounts)
save(myMycCounts, file = "data/MycCounts.RData")

3. DESeq2
为了评估跨细胞系的 ChIPseq 信号变化,我们将使用 DESeq2 包。
DESeq2 包包含一个工作流程,用于评估复制条件之间片段/读取丰度的局部变化。此工作流程包括标准化、方差估计、离群值移除/替换以及适用于高通量测序数据(即整数计数)的显着性测试。
要使用 DESeq2 工作流程,我们必须首先创建一个感兴趣条件的 data.frame,并将行名设置为我们的 BAM 文件名。
metaDataFrame <- data.frame(CellLine = c("Ch12", "Ch12", "Mel", "Mel"))
rownames(metaDataFrame) <- colnames(myMycCounts)
metaDataFrame

我们可以使用 DESeqDataSetFromMatrix() 函数来创建 DESeq2 对象。
我们必须将我们的计数矩阵提供给 countData 参数,我们的元数据 data.frame 提供给 colData 参数,并且我们在 rowRanges 的可选参数中包含我们可以依靠的非冗余峰值集。
最后,我们在我们希望测试设计参数的元数据 data.frame 中提供列的名称。
library(DESeq2)
deseqMyc <- DESeqDataSetFromMatrix(countData = assay(myMycCounts), colData = metaDataFrame,
design = ~CellLine, rowRanges = HC_Peaks)
我们现在可以使用 DESeq() 函数在我们的 DESeq2 对象上运行 DESeq2 工作流程。
deseqMyc <- DESeq(deseqMyc)

我们的 DESeq2 对象已更新,以包含有用的统计信息,例如我们的标准化值和每个非冗余峰调用中的信号方差。
deseqMyc

我们可以使用 results() 函数提取差异区域的信息。
我们向 results() 函数提供 DESeq2 对象、对对比参数感兴趣的比较以及返回格式参数的输出类型。
与对比参数的比较作为长度为 3 的向量提供,包括感兴趣的元数据列和要测试的组。
我们可以使用 order() 函数按 pvalue 对结果进行排序,以按最显着的变化进行排名。
MelMinusCh12 <- results(deseqMyc, contrast = c("CellLine", "Mel", "Ch12"), format = "GRanges")
MelMinusCh12 <- MelMinusCh12[order(MelMinusCh12$pvalue), ]
class(MelMinusCh12)

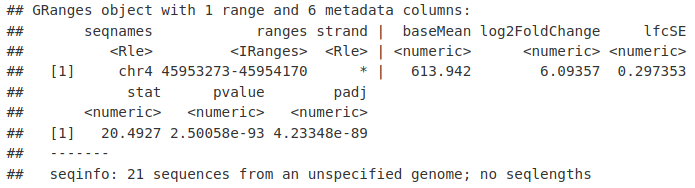
GRanges 对象包含有关在 DESeq2 中进行的比较的信息。
最有用的是它包含 IP 信号的差异,如 log2FoldChange 中的 log2 倍变化、pvalue 列中变化的重要性以及调整后的 p 值以解决 padj 列中的多重校正。
MelMinusCh12[1, ]
我们现在可以通过过滤 log2FoldChange 和 padj(针对多重校正调整的 p 值)小于 0.05,将我们的非冗余峰过滤为 Mel 或 Ch12 细胞系中信号明显更多的峰。
MelMinusCh12Filt <- MelMinusCh12[!is.na(MelMinusCh12$pvalue) | !is.na(MelMinusCh12$padj)]
UpinMel <- MelMinusCh12[MelMinusCh12$padj < 0.05 & MelMinusCh12$log2FoldChange >
0]
DowninMel <- MelMinusCh12[MelMinusCh12$padj < 0.05 & MelMinusCh12$log2FoldChange <
0]
export.bed(UpinMel, "UpinMel.bed")
export.bed(DowninMel, "DowninMel.bed")
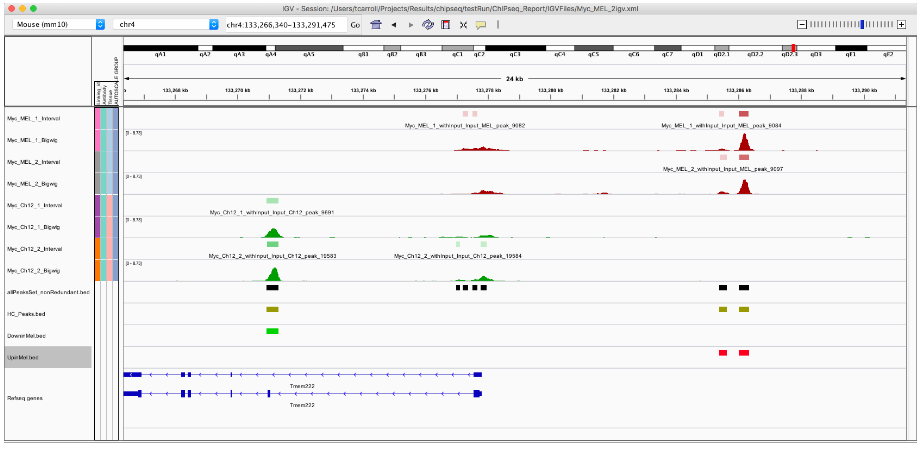
最后,我们可以使用 tracktables 包让我们在 IGV 中审查站点更容易一些。
tracktables 包的 makebedtable() 函数接受一个 GRanges 对象并编写一个包含 IGV 链接的 HTML 报告。
library(tracktables)
myReport <- makebedtable(MelMinusCh12Filt, "MelMinusCh12.html", basedirectory = getwd())
browseURL(myReport)








 苏公网安备32011502012024号
苏公网安备32011502012024号