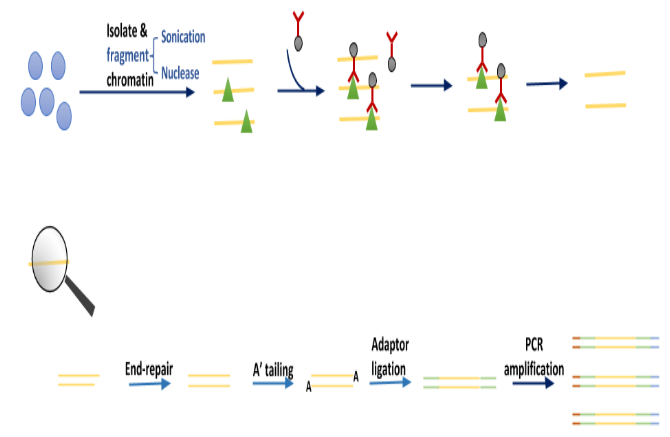
1. 片段长度评估
片段长度的预测是 ChIPseq 的重要组成部分,它会影响峰识别、峰识别和覆盖概况。
使用互相关或交叉覆盖可以评估按链进行的读取聚类,从而衡量质量。
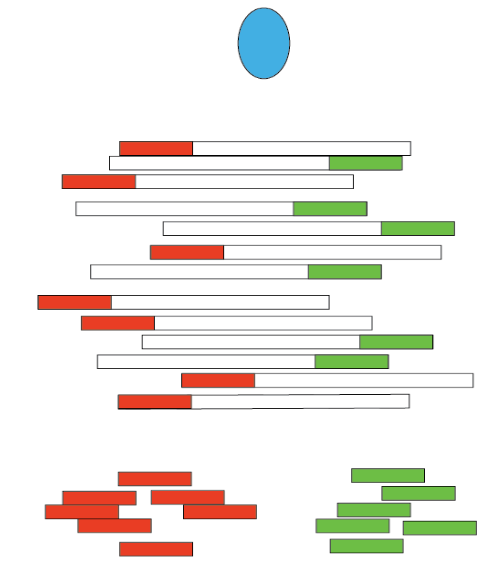
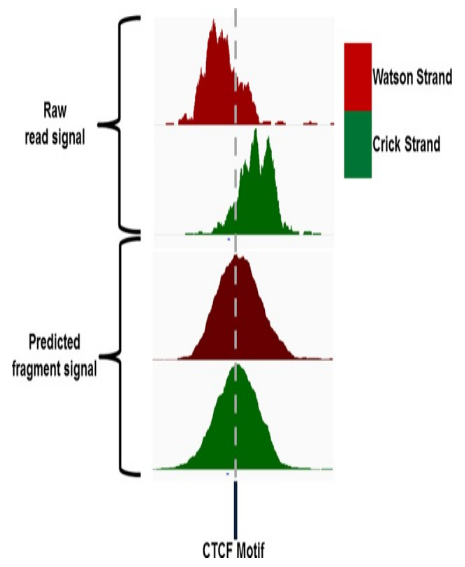
- 在 ChIPseq 中,通常是 dsDNA 的短单端读取。
- 片段的 5' 将在“+”链上测序
- 片段末端的 3' 将位于“-”链上。
- 虽然我们只有部分链序列,但根据预测的片段长度,我们可以预测整个片段
- “+”读数应仅在正方向延伸
- “-”只读负数
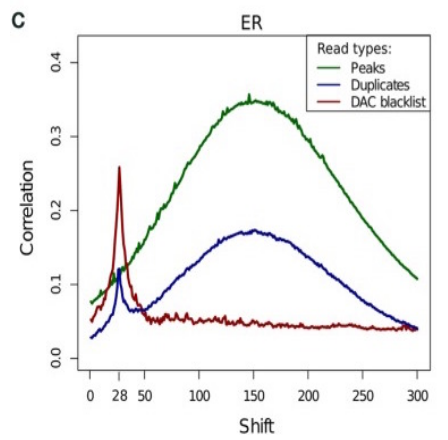
2. 交叉覆盖图
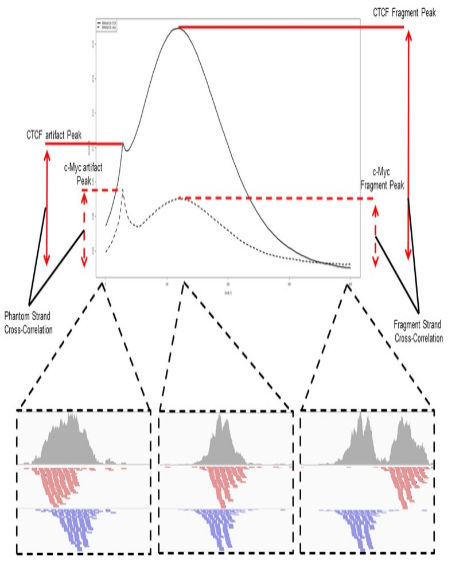
plotCC 函数可用于绘制我们的交叉覆盖图, plotCC() 函数接受我们的 ChIPQC 样本对象列表和一个 facetBy 参数,以允许我们对交叉覆盖配置文件进行分组。
plotCC(myQC, facetBy = "Sample")
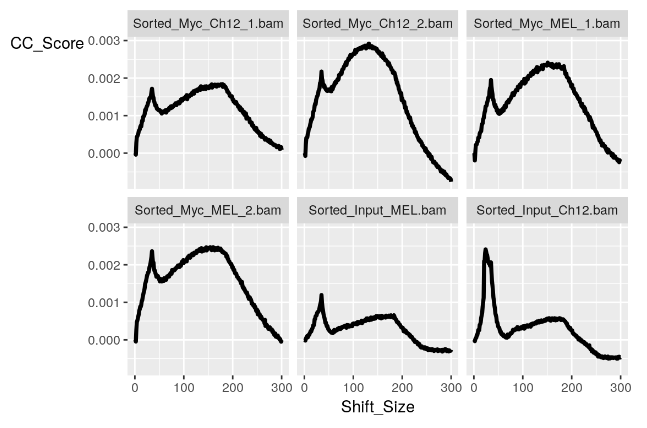
我们可以将元数据包含为 data.frame,其中第一列是我们的样本名称,以允许我们以不同的方式对我们的图进行分组。
myMeta <- data.frame(Sample = names(myQC), Tissue = c("Ch12", "Ch12", "MEL", "MEL",
"MEL", "Ch12"), Antibody = c(rep("Myc", 4), rep("Input", 2)))
myMeta

我们现在可以将我们的元数据包含到 addMetaData 参数中,这将允许我们对提供的元数据列进行 facetBy。
此外,我们在这里使用 colourBy 参数为抗体组添加颜色。
plotCC(myQC, facetBy = "Tissue", addMetaData = myMeta, colourBy = "Antibody")
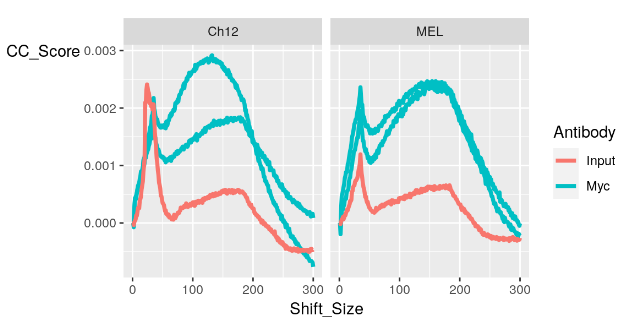
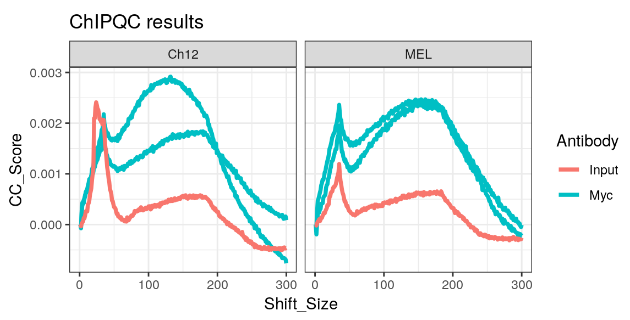
ChIPQC 中的所有图实际上都是在 ggplot2 中构建的,因此我们可以像所有 ggplot 对象一样编辑和更新我们的图。
plotCC(myQC, facetBy = "Tissue", addMetaData = myMeta, colourBy = "Antibody") + theme_bw() +
ggtitle("ChIPQC results")
3. 黑名单和SSD
3.1. 黑名单
ChIPseq 通常会显示常见伪影的存在,例如超高信号区域。这些区域可能会混淆峰识别、片段长度估计和 QC 指标。 Anshul Kundaje 创建了 DAC 黑名单作为参考,以帮助处理这些地区。
3.2. SSD
SSD 是其中一种对列入黑名单的工件敏感的措施。 SSD 是衡量整个基因组信号标准偏差的指标,较高的分数反映出大量的读数堆积。因此,SSD 可用于评估超高信号的范围和信号。但首先必须删除列入黑名单的区域。
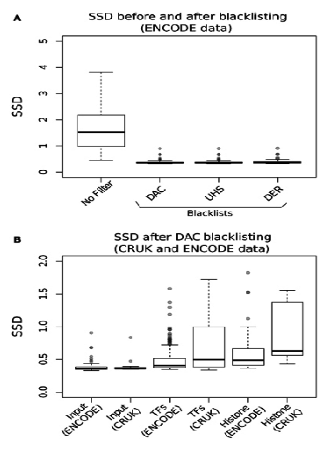
ChIPQC 在移除来自黑名单区域的信号之前和之后计算 SSD。plotSSD() 函数以红色绘制样本的黑名单前分数,以蓝色绘制黑名单后分数。
预先列入黑名单的 SSD 的较高分数可以表明该样本的黑名单区域中有很强的背景信号。
plotSSD(myQC) + xlim(0, 5)
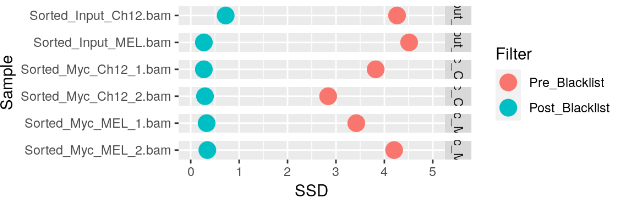
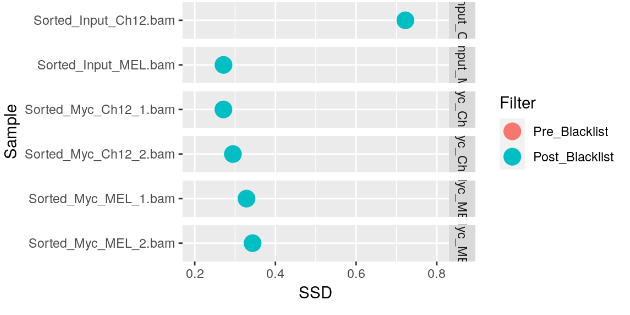
由于 SSD 分数受到黑名单的强烈影响,因此可能需要更改轴以查看黑名单后分数样本之间的任何差异。
更高的列入黑名单后的 SSD 分数反映了具有更强峰值信号的样本。
plotSSD(myQC) + xlim(0.2, 0.8)








 苏公网安备32011502012024号
苏公网安备32011502012024号