
引言
本系列将开展全新的转录组分析专栏,主要针对使用 DESeq2时可能出现的问题和方法进行展开。
SummarizedExperiment 输入
若您已经创建或获取了一个“SummarizedExperiment”对象,可以直接将其导入DESeq2进行分析。操作步骤如下:首先,需要导入包含气道数据集的相应程序包。
library("airway")
data("airway")
se <- airway
下面的构造函数示例说明了如何从“RangedSummarizedExperiment”对象“se”创建一个“DESeqDataSet”实例。
library("DESeq2")
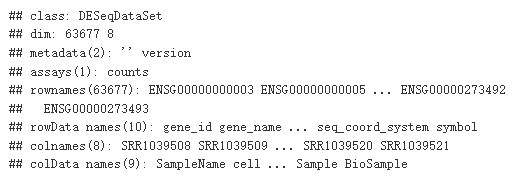
ddsSE <- DESeqDataSet(se, design = ~ cell + dex)
ddsSE
预筛选
尽管在执行DESeq2分析前,预筛选低表达量的基因并非必须,但预筛选有两个优点:移除读数极少的基因可以减少dds数据对象占用的内存,并加快DESeq2中计数模型的构建速度。此外,预筛选还能提升图形的可读性,因为那些在差异表达分析中没有信息量的特征不会出现在离散度图或MA图中。
在这里,进行预筛选,仅保留在至少一定数量样本中计数达到至少10的基因。对于常规RNA-seq数据,10是一个合理的阈值。建议的最小样本数量是最小的组大小,例如,这里的3个处理样本。如果没有明确的分组,可以选择一个非零计数有意义的最小样本数。当然,也可以跳过预筛选步骤,直接依赖于results()函数中提供的独立筛选方法,如IHW或genefilter。
smallestGroupSize <- 3
keep <- rowSums(counts(dds) >= 10) >= smallestGroupSize
dds <- dds[keep,]
关于因子水平的说明
通常情况下,R会按照字母顺序自动为因子指定一个参考水平。如果你没有明确告知DESeq2函数你想要比较的具体是哪个水平(比如,哪个水平代表对照组),那么比较将会基于这些水平的字母顺序来进行。这里有两种解决办法:你可以使用contrast参数明确指定results函数应该进行哪种比较(稍后将展示如何操作),或者你可以手动设定因子的水平。如果你希望在结果名称中看到参考水平变化的反映,你需要在调整水平之后运行DESeq或nbinomWaldTest/nbinomLRT。设定因子水平可以通过两种方法实现,一种是通过factor函数:
dds$condition <- factor(dds$condition, levels = c("untreated","treated"))
…或者使用 relevel,只需指定参考级别:
dds$condition <- relevel(dds$condition, ref = "untreated")
当你需要对“DESeqDataSet”对象的列进行筛选,例如在分析中排除某些样本时,可能会遇到一个或多个设计公式中变量的水平对应的所有样本都被移除的情形。这时,可以利用“droplevels”函数来删除那些在当前“DESeqDataSet”中已经没有样本对应的水平。
dds$condition <- droplevels(dds$condition)
合并技术重复
DESeq2 提供了一个名为 collapseReplicates 的功能,它可以帮助将技术重复的计数数据整合到计数矩阵的单一列中。所谓“技术重复”,指的是对同一样本库进行多次测序。注意,不应使用此功能合并生物学重复样本。
帕西拉数据集简介
继续使用上述通过计数矩阵方法构建的帕西拉(pasilla)数据集。这个数据集来源于对果蝇(Drosophila melanogaster)细胞培养的实验,旨在探究 RNAi 技术敲除剪接因子帕西拉(pasilla)的效果(Brooks 等,2011年)。
差异表达分析
差异表达分析的标准步骤被集成到了一个名为 DESeq 的函数中。
使用 results 函数来生成结果表,这个表包含了 log2 倍数变化、p 值和校正后的 p 值。如果没有给 results 函数提供额外的参数,那么 log2 倍数变化和 Wald 测试 p 值将默认针对设计公式中的最后一个变量,如果这个变量是一个因子,那么比较将是这个变量的最新水平与参考水平之间的比较(如前所述,关于因子水平)。不过,只要用户明确指定了要比较的变量,设计中的变量顺序并不重要。
在结果表上方的控制台中,会直接打印出比较的详细信息。例如,“条件 处理 vs 未处理”这样的文本表明,这里估计的是处理组与未处理组之间的对数倍数变化(log2(处理/未处理))。
dds <- DESeq(dds)
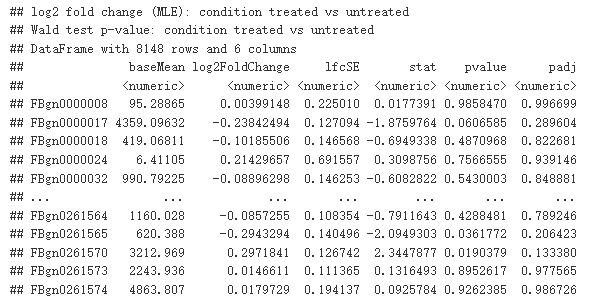
res <- results(dds)
res
请注意,可以使用以下任一等效命令指定要为其构建结果表的系数或对比度:
res <- results(dds, name="condition_treated_vs_untreated")
res <- results(dds, contrast=c("condition","treated","untreated"))
这两个命令等价性的一个例外情况是,当使用 contrast 参数时,如果在两组比较中,所有计数都为零(而其他组有非零计数),那么这两组的估计对数倍数变化(LFC)会被额外设置为0。如果希望在这些情况下将LFC设置为0,可以使用contrast来构建结果表。
对数倍数变化的收缩对于基因的可视化和排序非常有用。为了实现LFC的收缩,将dds对象传递给lfcShrink函数。在这里,指定使用apeglm方法来收缩效应大小(LFC估计)。
提供了dds对象以及希望收缩的系数的名称或编号,其中编号指的是该系数在resultsNames(dds)中出现的顺序。
resultsNames(dds)
## [1] "Intercept" "condition_treated_vs_untreated"
resLFC <- lfcShrink(dds, coef="condition_treated_vs_untreated", type="apeglm")
resLFC









 苏公网安备32011502012024号
苏公网安备32011502012024号