在下部分中,我们将研究如何使用 R/Bioconductor 识别开放区域中的变化。
在这里,我们将采用类似于 Diffbind 中的方法,并在 ATACseq 分析中合理建立。
1. 识别非冗余峰
首先,我们将定义至少 2 个样本中存在的一组非冗余峰,并使用这些峰使用 DESeq2 评估无核小体 ATACseq 信号的变化。在这里,我们使用与 ChIPseq 相同的方法来推导差异的一致峰。
我们在所有样本中取峰并将它们减少为一组非冗余峰。然后我们可以在每个样本上创建这些峰存在/不存在的矩阵。
peaks <- dir("~/Downloads/ATAC_Workshop/ATAC_Data/ATAC_Peaks_forCounting/", pattern = "*.narrowPeak",
full.names = TRUE)
myPeaks <- lapply(peaks, ChIPQC:::GetGRanges, simple = TRUE)
allPeaksSet_nR <- reduce(unlist(GRangesList(myPeaks)))
overlap <- list()
for (i in 1:length(myPeaks)) {
overlap[[i]] <- allPeaksSet_nR %over% myPeaks[[i]]
}
overlapMatrix <- do.call(cbind, overlap)
colnames(overlapMatrix) <- basename(peaks)
mcols(allPeaksSet_nR) <- overlapMatrix
allPeaksSet_nR[1:2, ]

我们在测试之前过滤掉黑名单和 ChrM 中的峰值,以消除潜在的伪差调用。
blklist <- import.bed("data/ENCFF547MET.bed.gz")
nrToCount <- allPeaksSet_nR[!allPeaksSet_nR %over% blklist & !seqnames(allPeaksSet_nR) %in%
"chrM"]
nrToCount
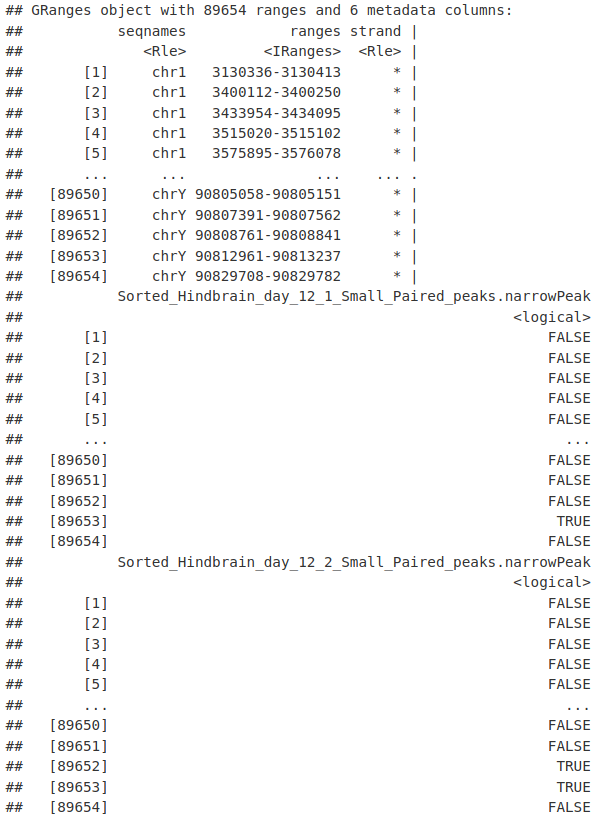
2. 差异计数
我们现在确定出现非冗余峰的样本数量。在这里,我们将 rowSums() 函数与我们的出现矩阵一起使用,并选择出现在至少 2 个样本中的那些样本。
library(Rsubread)
occurrences <- rowSums(as.data.frame(elementMetadata(nrToCount)))
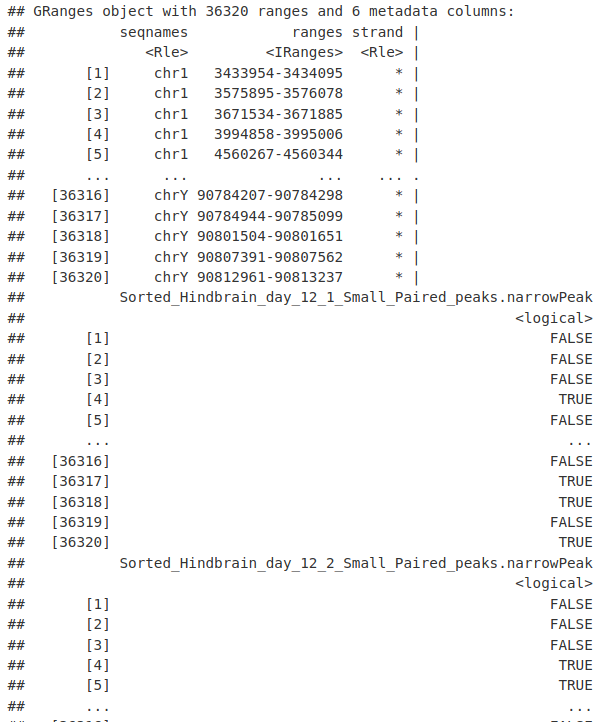
nrToCount <- nrToCount[occurrences >= 2, ]
nrToCount
现在我们必须设置要计数的区域,我们可以使用 summariseOverlaps() 来计算到达峰值的成对读数,就像我们对 ChIPseq 所做的那样。
我们必须调整双端读取的计数,因此我们另外将 singleEnd 参数设置为 FALSE。
library(GenomicAlignments)
bamsToCount <- dir("~/Downloads/ATAC_Workshop/ATAC_Data/ATAC_BAM_forCounting/", full.names = TRUE,
pattern = "*.\\.bam$")
myCounts <- summarizeOverlaps(consensusToCount, bamsToCount, singleEnd = FALSE)
colnames(myCounts) <- c("HindBrain_1", "HindBrain_2", "Kidney_1", "Kidney_2", "Liver_1",
"Liver_2")
3. DESeq2
有了我们在无核小体区域中的片段计数,我们现在可以构建一个 DESeq2 对象。
我们将计数区域的 GRanges 传递给 DESeqDataSetFromMatrix 函数,以便稍后从 DESeq2 访问这些区域。
library(DESeq2)
load("data/myCounts.RData")
Group <- factor(c("HindBrain", "HindBrain", "Kidney", "Kidney", "Liver", "Liver"))
metaData <- data.frame(Group, row.names = colnames(myCounts))
atacDDS <- DESeqDataSetFromMatrix(assay(myCounts), metaData, ~Group, rowRanges = rowRanges(myCounts))
atacDDS <- DESeq(atacDDS)

使用新的 DESeq2 对象,我们现在可以测试组间 ATACseq 信号的任何差异。在此示例中,我们将查看后脑样本和肾脏样本之间的差异。我们在这里返回一个 GRanges 对象,以允许我们执行更多的 GenomicRanges 操作。
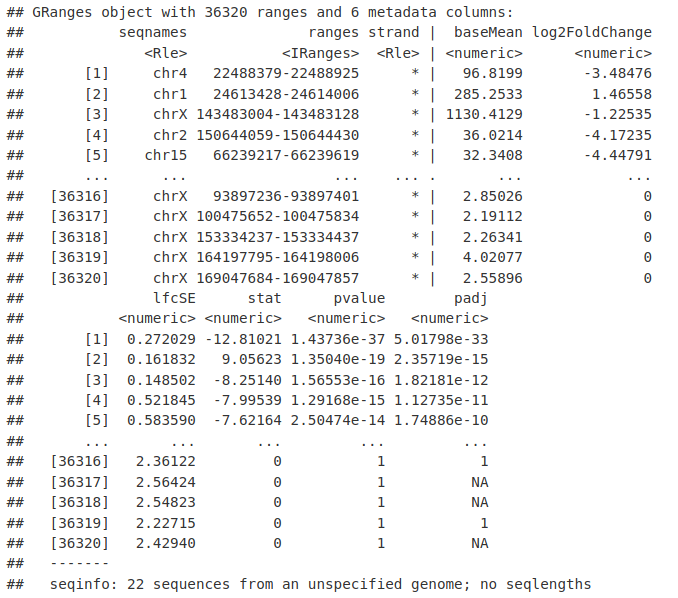
KidneyMinusHindbrain <- results(atacDDS, c("Group", "Kidney", "HindBrain"), format = "GRanges")
KidneyMinusHindbrain <- KidneyMinusHindbrain[order(KidneyMinusHindbrain$pvalue)]
KidneyMinusHindbrain
我们可以子集化为仅在启动子内打开区域,然后使用 tracktables 包中的 makebedtable() 函数创建一个 HTML 表以查看 IGV 中的结果。

library(TxDb.Mmusculus.UCSC.mm10.knownGene)
toOverLap <- promoters(TxDb.Mmusculus.UCSC.mm10.knownGene, 500, 500)
KidneyMinusHindbrain <- KidneyMinusHindbrain[(!is.na(KidneyMinusHindbrain$padj) &
KidneyMinusHindbrain$padj < 0.05) & KidneyMinusHindbrain %over% toOverLap, ]
myReport <- makebedtable(KidneyMinusHindbrain, "KidneyMinusHindbrain.html", getwd())
browseURL(myReport)
4. 差异注释
在最后一部分,我们可以将我们的差异 ATACseq 区域注释到基因,然后使用基因信息来测试 GO 集的富集。
由于我们有 TSS +/- 500bp 范围内的区域子集,此时我们可以使用标准富集分析。这里我们使用clusterProfiler来识别富集。
anno_KidneyMinusHindbrain <- annotatePeak(KidneyMinusHindbrain, TxDb = TxDb.Mmusculus.UCSC.mm10.knownGene,
verbose = FALSE)
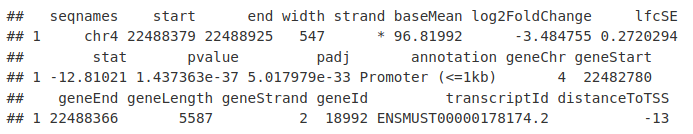
DB_ATAC <- as.data.frame(anno_KidneyMinusHindbrain)
DB_ATAC[1, ]
由于我们有 TSS +/- 500bp 范围内的区域子集,此时我们可以使用标准富集分析。这里我们使用 clusterProfiler 来识别富集。
library(clusterProfiler)
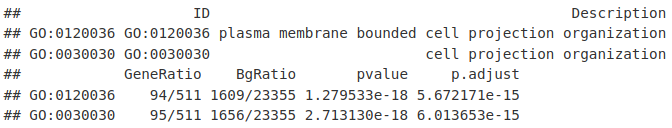
go <- enrichGO(DB_ATAC$geneId, OrgDb = "org.Mm.eg.db", ont = "BP", maxGSSize = 5000)
go[1:2, 1:6]








 苏公网安备32011502012024号
苏公网安备32011502012024号