首先介绍一下什么是ATAC-seq。我们最常见的就是RNA-seq的测序,那么ATAC-seq是干什么的呢?其实我们知道RNA的转录受到调控元件的控制(例如增强子),那么如何来获得这些调控元件呢,就是依靠这个ATAC-seq测序,那么为什么ATAC-seq为什么能够获得这些调控元件呢?因为ATAC-seq使用了一种酶(Tn5),这种酶能够切割DNA的开放区域;因为DNA是被包裹成核小体的形式的,所以要发挥作用需要变成开放形式才可以有转录因子能够结合上去,如下图:
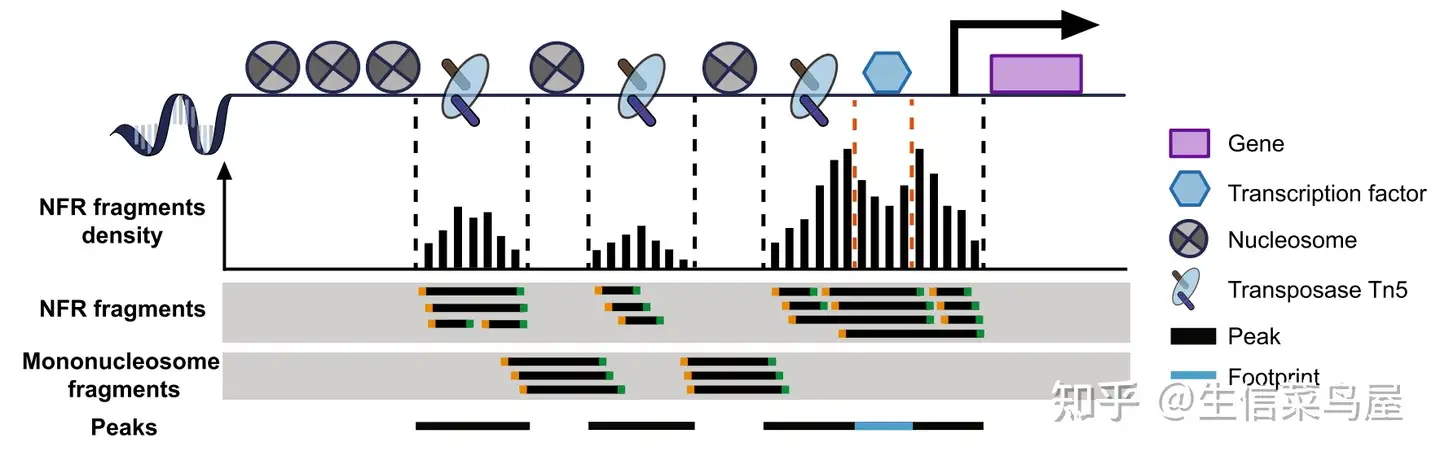
转录因子结合在染色质的开放区,调控基因表达[1]。
现在大家明白ATAC-seq的作用了吧,总结来说:就是ATAC-seq是对染色质的开放区域进行测序,而这些开放区域可能会对某些基因进行调控。
接下来就是ATAC-seq的数据分析了。
首先拿到fq文件之后,我们首先需要对其进行过滤:
fastp -i fq1 -I fq2 -o out1 -O out2 -w 16
建议大家使用fastp的默认参数,因为ATAC-seq的片段长度只有50bp左右。因此很多公司给的clean data是150bp的话是不合理的,可能会导致很多信息被漏掉(这是我掉的坑)。
拿到过滤后的fq文件之后,需要把其比对到基因组上,因为是不考虑剪切比对,所以我们使用bowtie2:
bowtie2 -p 20 -x /sdd/xujiahao/chip/bowtie2_index_mm10/mm10 -1 $fq1 -2 $fq2 -X 2000 -S ${name}.sam &> ${name}.txt
-X:表示片段长度最大是多少,因为ATAC-seq在600bp的时候几乎就没有了,因此2000是足够了。
比对完之后,就需要进行过滤:
samtools view -Su ${name}.sam -@ 8 -o ${name}.bam ####把sam文件转为bam文件
samtools view -F 1804 -f 2 -q 30 -u $id -o ${name}.1830.bam -@ 10 ###过滤孤立片段以及比对质量低于30
samtools sort -n ${name}.1830.bam -o ${name}.1830.ns.bam -@ 10 ####对过滤后的bam按照name排序
samtools fixmate -r ${name}.1830.ns.bam ${name}.1830.ns.fm.bam -@ 10 ###对reads进行配对
samtools view -F 1804 -f 2 -u ${name}.1830.ns.fm.bam -@ 10 | samtools sort /dev/stdin -o ${name}.filt.bam -@ 8 ####过滤配对后的孤立片段
sambamba markdup ${name}.filt.bam ${name}.md.bam -t 8 ###标记重复片段
samtools view -F 1804 -f 2 -b ${name}.md.bam -o ${name}.d.bam -@ 10 ####过滤重复片段
在此我们就获得了干净的bam文件。
之后则需要进行ATAC-seq特征指标进行QC,这个在下期讲解。 | 




 苏公网安备32011502012024号
苏公网安备32011502012024号