1. 注释开放区域
将已识别的无核小体区域与基因组特征(如基因和增强子)相关联通常很有趣。
一旦注释到基因或增强子的基因,我们就可以开始将 ATACseq 数据与这些基因的特征相关联。 (功能注释、表达变化、其他表观遗传状态)。
2. 基因注释
将无核小体区域注释到基因的一种简单方法是将区域与其最近的基因或在基因转录起始位点周围的窗口内相关联。我们可以使用 chipseeker 库来识别最接近我们区域的基因,并为我们提供此注释的简单摘要和可视化。
我们使用来自 TxDb.Hsapiens.UCSC.hg19.knownGene 的基因模型并将其提供给 ChIPseeker 包 annotatePeak 函数。
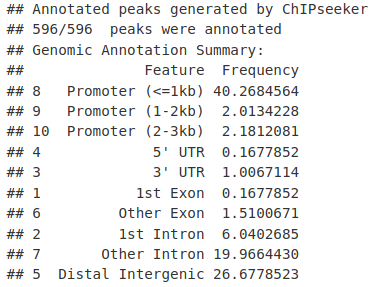
ChIPseeker 的 csAnno 对象随后将显示基因组区域中峰值百分比的细分。
library(ChIPseeker)
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
MacsCalls_Anno <- annotatePeak(MacsCalls, TxDb = TxDb.Hsapiens.UCSC.hg19.knownGene)

MacsCalls_Anno
3. 注释分布
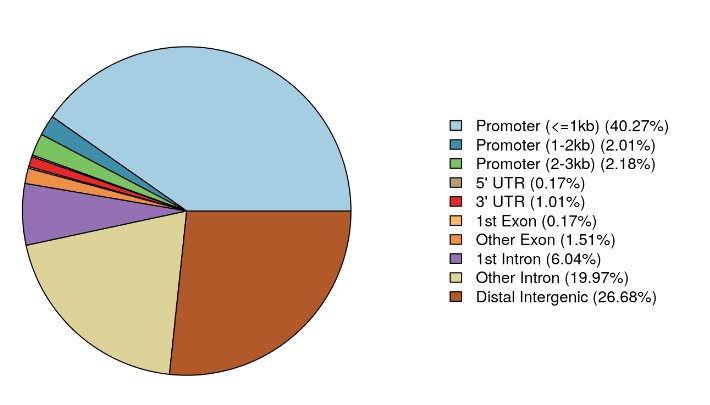
除了向我们展示注释分布表之外,我们还可以使用 plotAnnoPie 和 plotAnnoBar 函数将其可视化。
plotAnnoPie(MacsCalls_Anno)
4. 注释无核小体区域
有了这些信息,我们就可以将我们的 peaks/nuc 自由区域子集化为那些只在 TSS 区域着陆的区域 (+/- 500)。
MacsGR_Anno <- as.GRanges(MacsCalls_Anno)
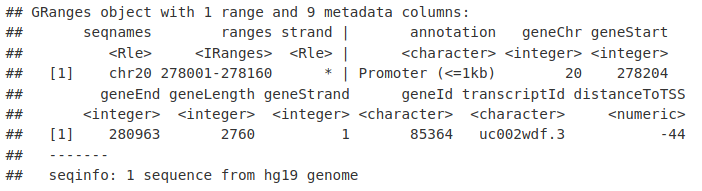
MacsGR_TSS <- MacsGR_Anno[abs(MacsGR_Anno$distanceToTSS) < 500]
MacsGR_TSS[1, ]
5. 无核小体区域功能分析
ATACseq 分析的另一个常见步骤是识别与无核小体区域相关的基因中的任何功能富集。
一种方法是采用我们从 ChIPseeker 中鉴定为具有无核小体区域的基因,并使用 GOseq 等标准工具测试这些基因的功能富集。我们可以使用 submitGreatJob 函数将峰值调用提交给 GREAT,并使用 availableCategories 查看可用的结果类别。
library(rGREAT)
great_Job <- submitGreatJob(MacsCalls, species = "hg19")
availableCategories(great_Job)

对于此示例,我们使用 getEnrichmentTables 函数选择 GO 类别的结果表,然后查看生物过程的结果。
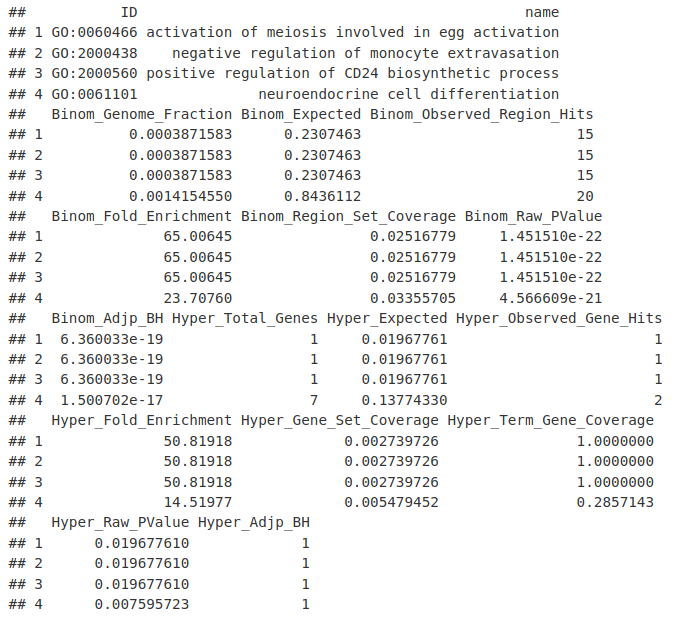
great_ResultTable = getEnrichmentTables(great_Job, category = "GO")
names(great_ResultTable)

great_ResultTable[["GO Biological Process"]][1:4, ]








 苏公网安备32011502012024号
苏公网安备32011502012024号