数据加载
在本案例中,将采用之前在轨迹构建案例中已经介绍并处理过的数据集。
library(Signac)
library(Seurat)
bone <- readRDS("cd34.rds")
DimPlot(bone, label = TRUE)
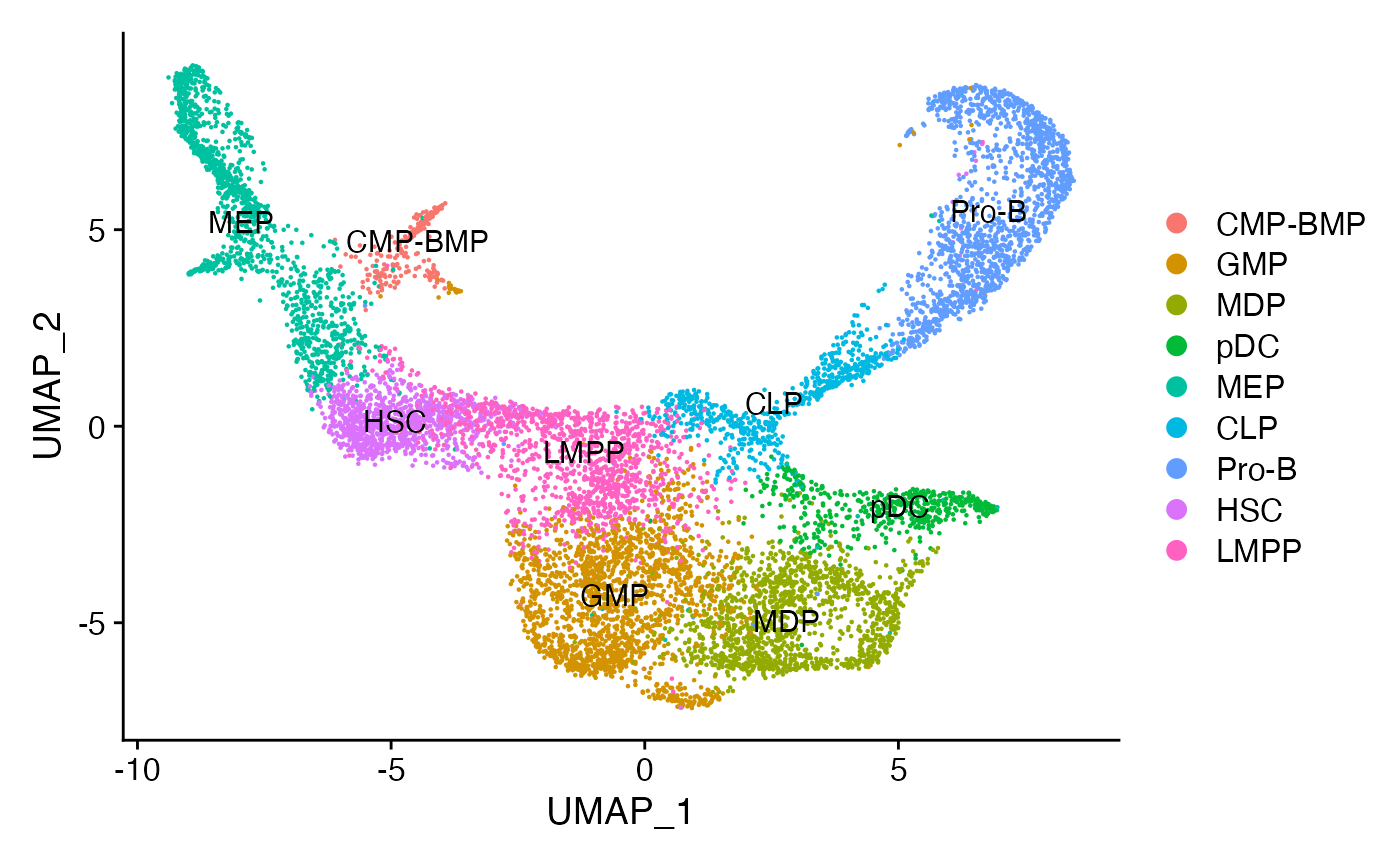
要执行足迹分析,必须首先向对象中添加Motif 信息,这包括每个Motif 的精确位置。这一过程可以通过使用“motif”和“packages”这两个包中的函数来实现。
library(motifmatchr)
library(JASPAR2020)
library(TFBSTools)
library(BSgenome.Hsapiens.UCSC.hg19)
# extract position frequency matrices for the motifs
pwm <- getMatrixSet(
x = JASPAR2020,
opts = list(species = 9606, all_versions = FALSE)
)
# add motif information
bone <- AddMotifs(bone, genome = BSgenome.Hsapiens.UCSC.hg19, pfm = pwm)
Motif 足迹分析
现在可以对任何已知位置信息的Motif 进行足迹分析。通常,这会涵盖基因组中所有该Motif 的实例。也可以设置 in.peaks = TRUE 参数,以便只考虑那些位于分析中峰值区域内的Motif 。Footprint() 函数会收集所有必要的数据,并将其保存在分析结果中。之后,可以使用 PlotFootprint() 函数来绘制这些基序的足迹图。
# gather the footprinting information for sets of motifs
bone <- Footprint(
object = bone,
motif.name = c("GATA2", "CEBPA", "EBF1"),
genome = BSgenome.Hsapiens.UCSC.hg19
)
# plot the footprint data for each group of cells
p2 <- PlotFootprint(bone, features = c("GATA2", "CEBPA", "EBF1"))
p2 + patchwork::plot_layout(ncol = 1)

| 







 苏公网安备32011502012024号
苏公网安备32011502012024号