引言
在这篇指南中,我们介绍了Seurat的一个新扩展功能,用以分析新型的空间解析数据,将重点介绍由不同成像技术生成的三个公开数据集。
- Vizgen MERSCOPE(用于小鼠大脑研究)
- Nanostring CosMx空间分子成像仪(用于FFPE人类肺组织)
- Akoya CODEX(用于人类淋巴结研究)
人肺:Nanostring CosMx Spatial Molecular Imager
这个数据集是通过Nanostring公司的CosMx空间分子成像仪(SMI)生成的。这种SMI技术能够进行多路复用的单分子分析,不仅可以检测RNA和蛋白质,还能够直接应用于固定石蜡包埋(FFPE)组织。数据集包含了从5个非小细胞肺癌(NSCLC)患者身上采集的8个FFPE样本,并且已经对公众开放下载。这个基因检测板涵盖了960种不同的转录本。
在本案例研究中,我们选取了8个样本中的一个(肺样本5,复制品1)进行加载。我们运用了LoadNanostring()函数来解析在公开下载站点上可以获取的数据。
对于这个数据集,我们并没有进行无监督分析,而是将Nanostring的分析结果与我们的Azimuth健康人类肺脏参考数据库进行对比,这个数据库是通过单细胞RNA测序(scRNA-seq)技术建立的。我们使用的是Azimuth软件的0.4.3版本以及人类肺脏参考数据库的1.0.0版本。你可以在指定的链接下载预先计算好的分析结果,这些结果包括了注释信息、预测分数以及UMAP的可视化图。每个细胞平均检测到的转录本数量是249,这在进行细胞注释时确实带来了一定的不确定性。
nano.obj <- LoadNanostring(data.dir = "/brahms/hartmana/vignette_data/nanostring/lung5_rep1", fov = "lung5.rep1")
# add in precomputed Azimuth annotations
azimuth.data <- readRDS("/brahms/hartmana/vignette_data/nanostring_data.Rds")
nano.obj <- AddMetaData(nano.obj, metadata = azimuth.data$annotations)
nano.obj[["proj.umap"]] <- azimuth.data$umap
Idents(nano.obj) <- nano.obj$predicted.annotation.l1
# set to avoid error exceeding max allowed size of globals
options(future.globals.maxSize = 8000 * 1024^2)
nano.obj <- SCTransform(nano.obj, assay = "Nanostring", clip.range = c(-10, 10), verbose = FALSE)
# text display of annotations and prediction scores
head(slot(object = nano.obj, name = "meta.data")[2:5])
## nCount_Nanostring nFeature_Nanostring predicted.annotation.l1
## 1_1 23 19 Dendritic
## 2_1 26 23 Macrophage
## 3_1 74 51 Neuroendocrine
## 4_1 60 48 Macrophage
## 5_1 52 39 Macrophage
## 6_1 5 5 CD4 T
## predicted.annotation.l1.score
## 1_1 0.5884506
## 2_1 0.5707920
## 3_1 0.5449661
## 4_1 0.6951970
## 5_1 0.8155319
## 6_1 0.5677324
我们可以可视化 Nanostring 细胞和注释,并将其投影到参考定义的 UMAP 上。请注意,对于此 NSCLC 样本,肿瘤样本被注释为“基础”,这是健康参考中最接近的细胞类型匹配。
DimPlot(nano.obj)
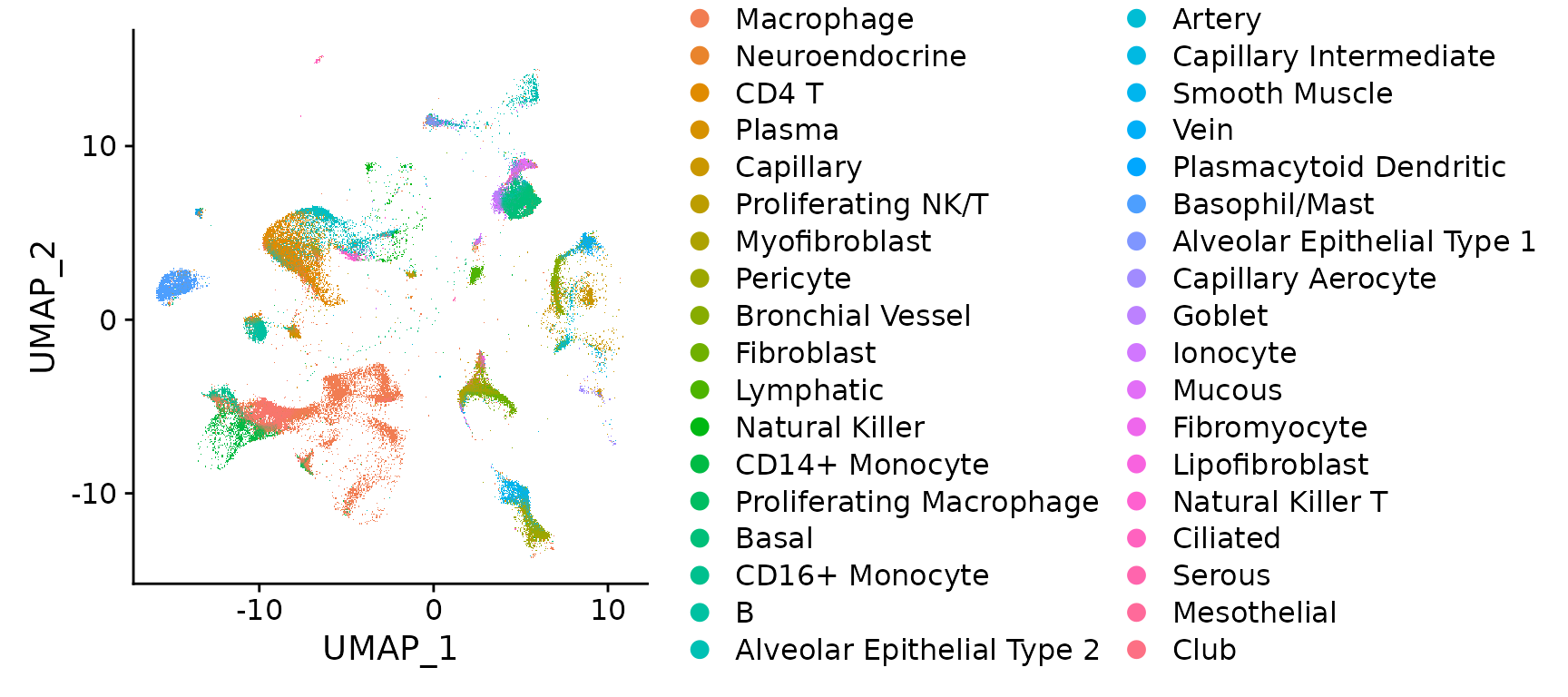
细胞类型和表达定位模式的可视化
正如之前的例子所展示的,ImageDimPlot() 这个函数会根据细胞在空间上的分布位置来绘制它们,并依据细胞被指定的类型来对它们进行颜色标记。可以观察到,基底细胞群(也就是肿瘤细胞)在空间上的排列非常紧凑有序,这与我们的预期是一致的。
ImageDimPlot(nano.obj, fov = "lung5.rep1", axes = TRUE, cols = "glasbey")
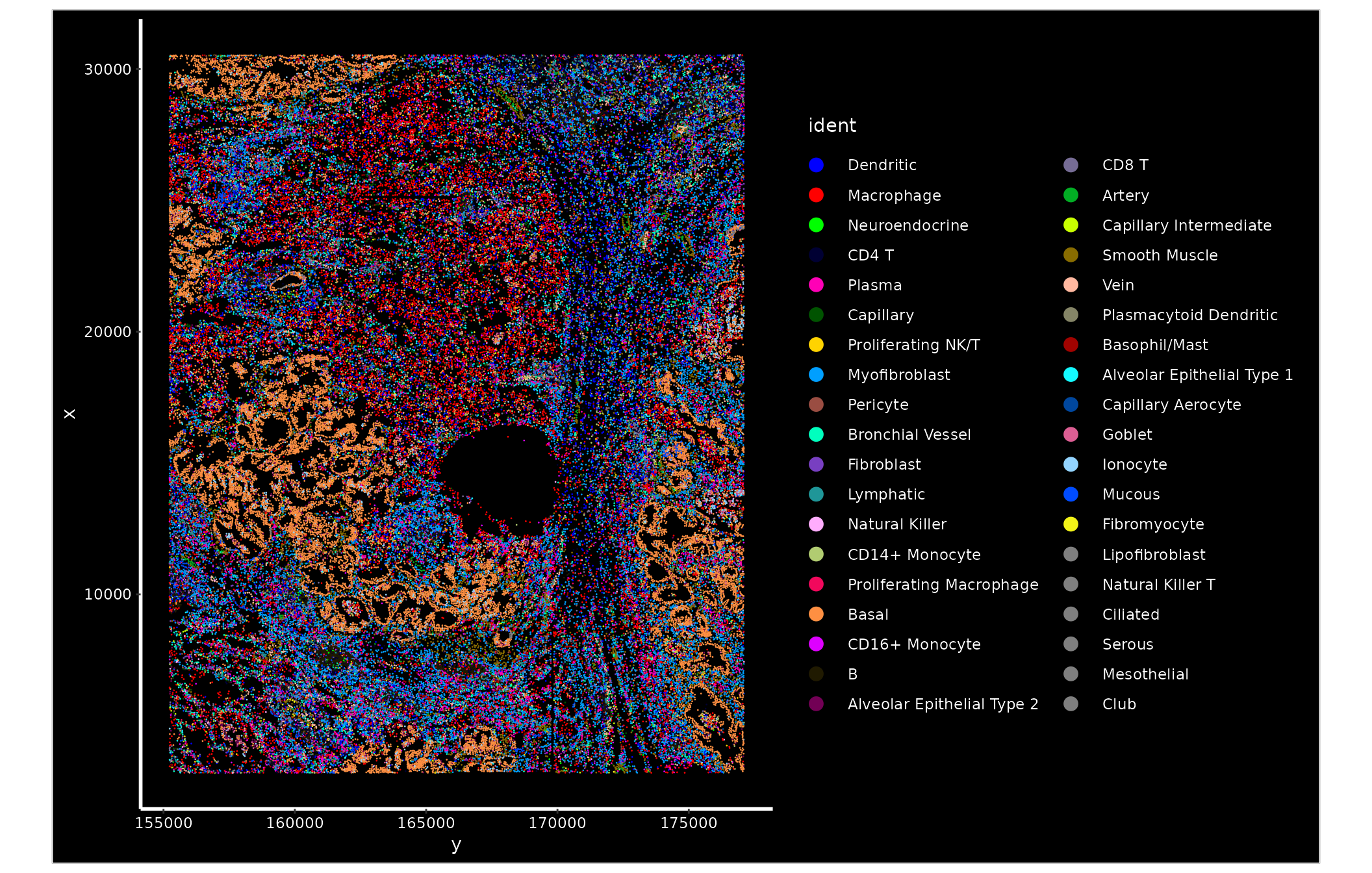
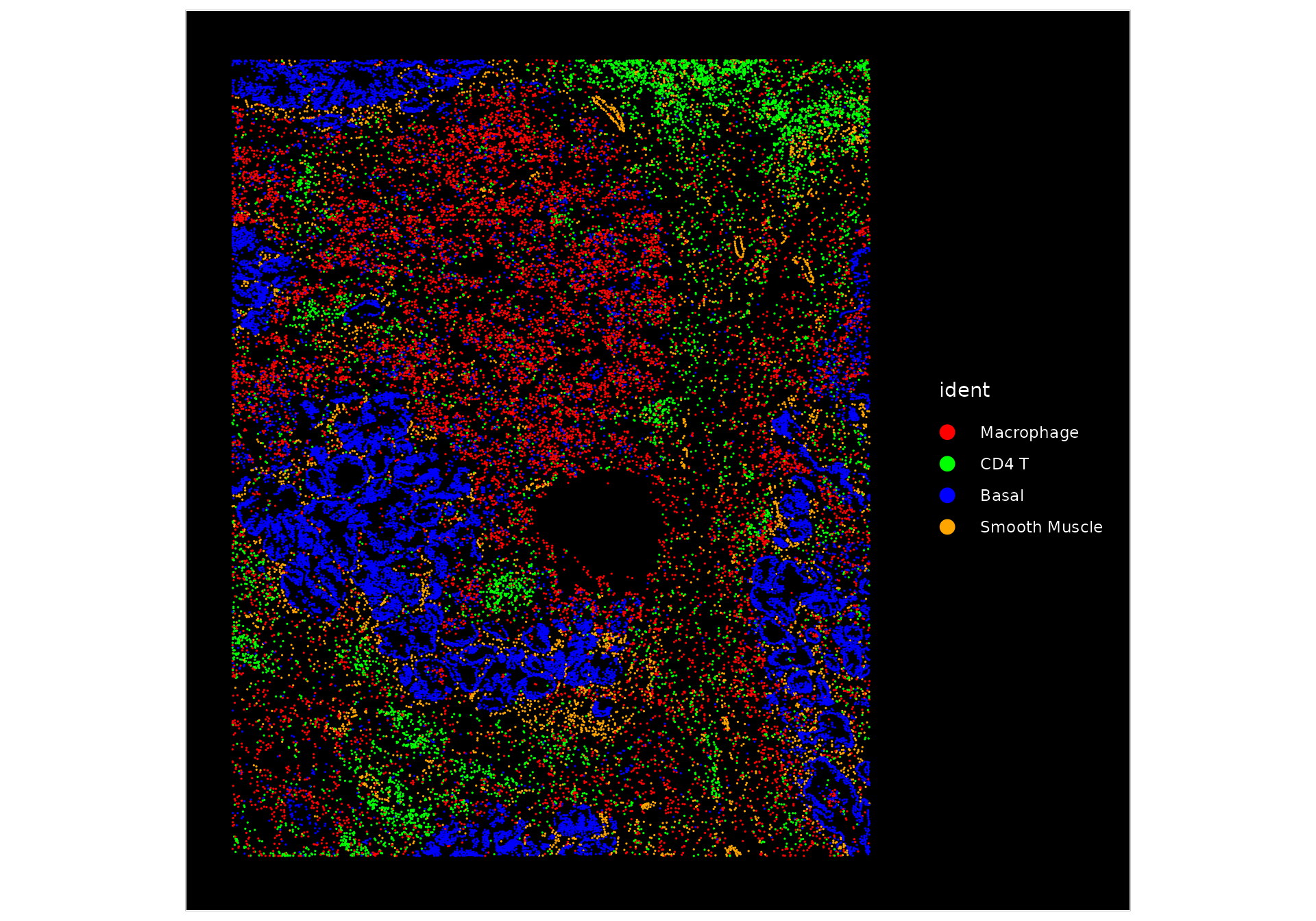
由于存在多种细胞类型,我们可以突出显示一些选定组的定位。
ImageDimPlot(nano.obj, fov = "lung5.rep1", cells = WhichCells(nano.obj, idents = c("Basal", "Macrophage",
"Smooth Muscle", "CD4 T")), cols = c("red", "green", "blue", "orange"), size = 0.6)
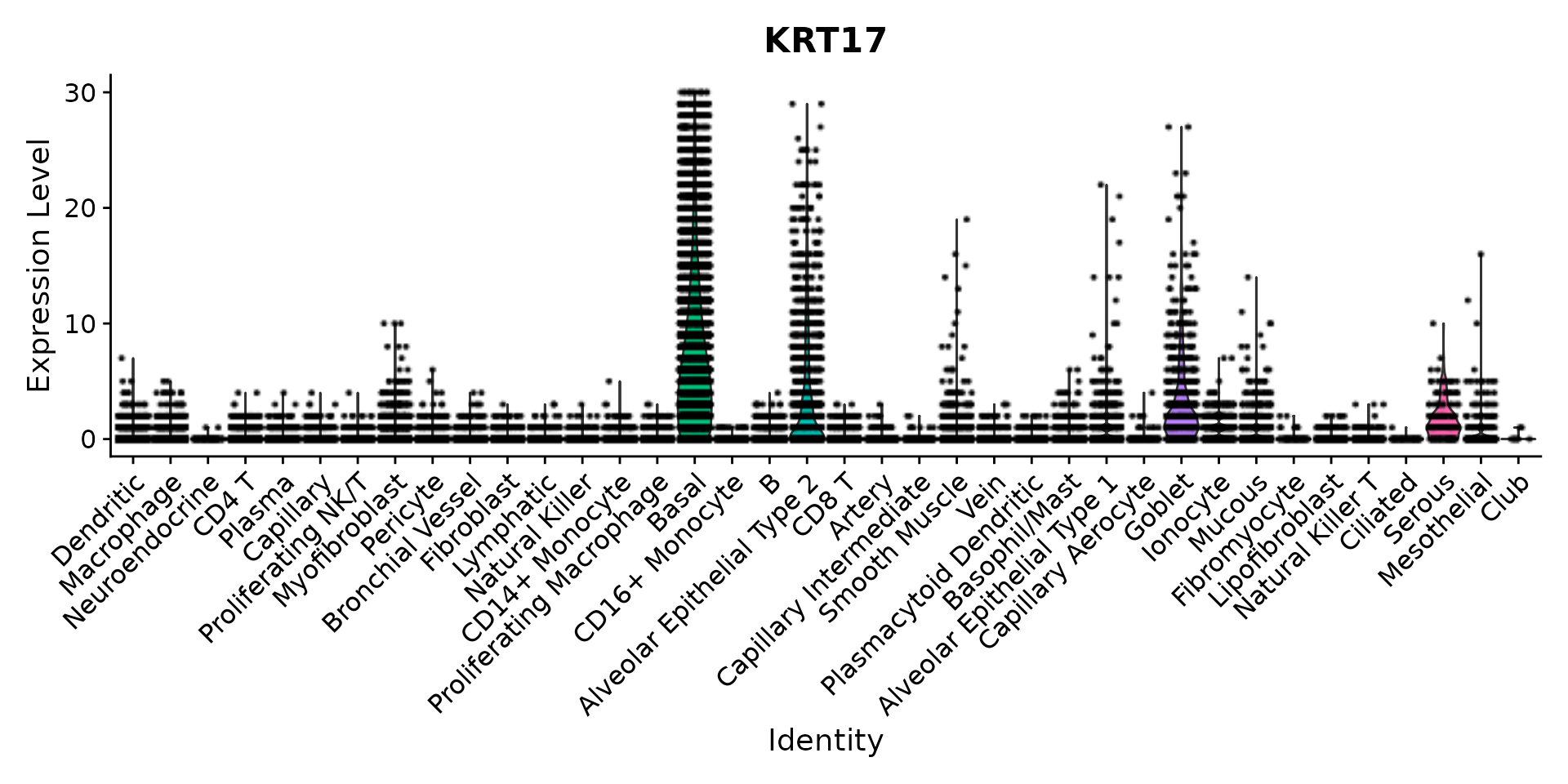
我们还可以通过几种不同的方式可视化基因表达标记:
VlnPlot(nano.obj, features = "KRT17", assay = "Nanostring", layer = "counts", pt.size = 0.1, y.max = 30) +
NoLegend()
FeaturePlot(nano.obj, features = "KRT17", max.cutoff = "q95")
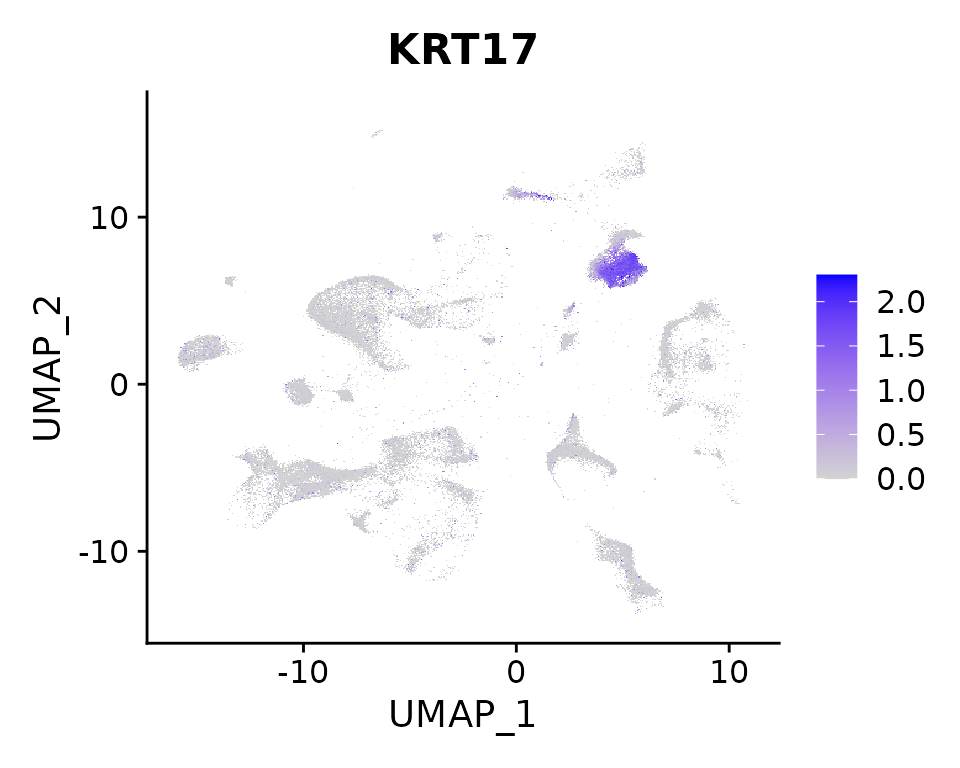
p1 <- ImageFeaturePlot(nano.obj, fov = "lung5.rep1", features = "KRT17", max.cutoff = "q95")
p2 <- ImageDimPlot(nano.obj, fov = "lung5.rep1", alpha = 0.3, molecules = "KRT17", nmols = 10000) +
NoLegend()
p1 + p2

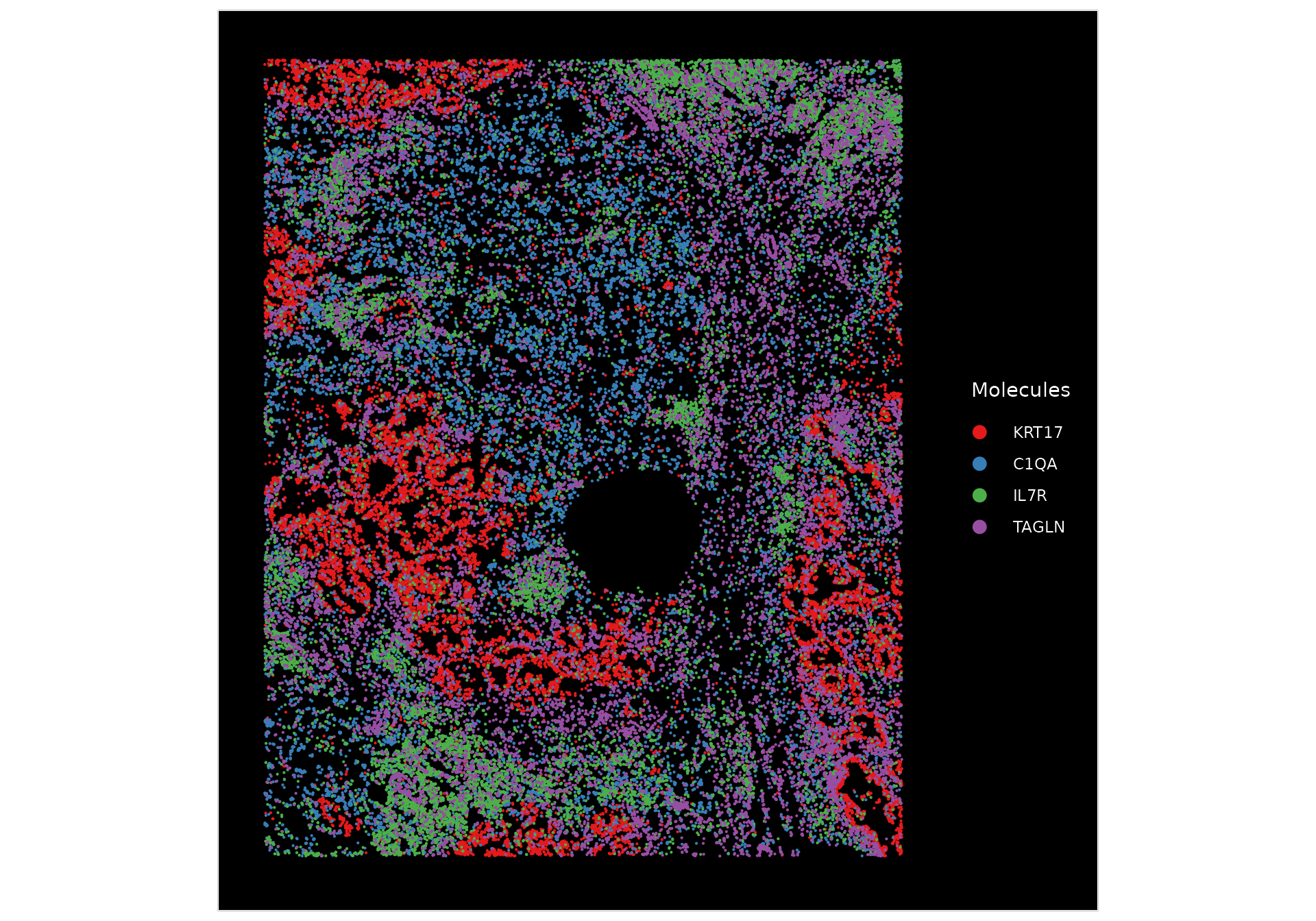
我们可以绘制分子图,以便共同可视化多个标记物的表达,包括 KRT17(基底细胞)、C1QA(巨噬细胞)、IL7R(T 细胞)和 TAGLN(平滑肌细胞)。
# Plot some of the molecules which seem to display spatial correlation with each other
ImageDimPlot(nano.obj, fov = "lung5.rep1", group.by = NA, alpha = 0.3, molecules = c("KRT17", "C1QA",
"IL7R", "TAGLN"), nmols = 20000)
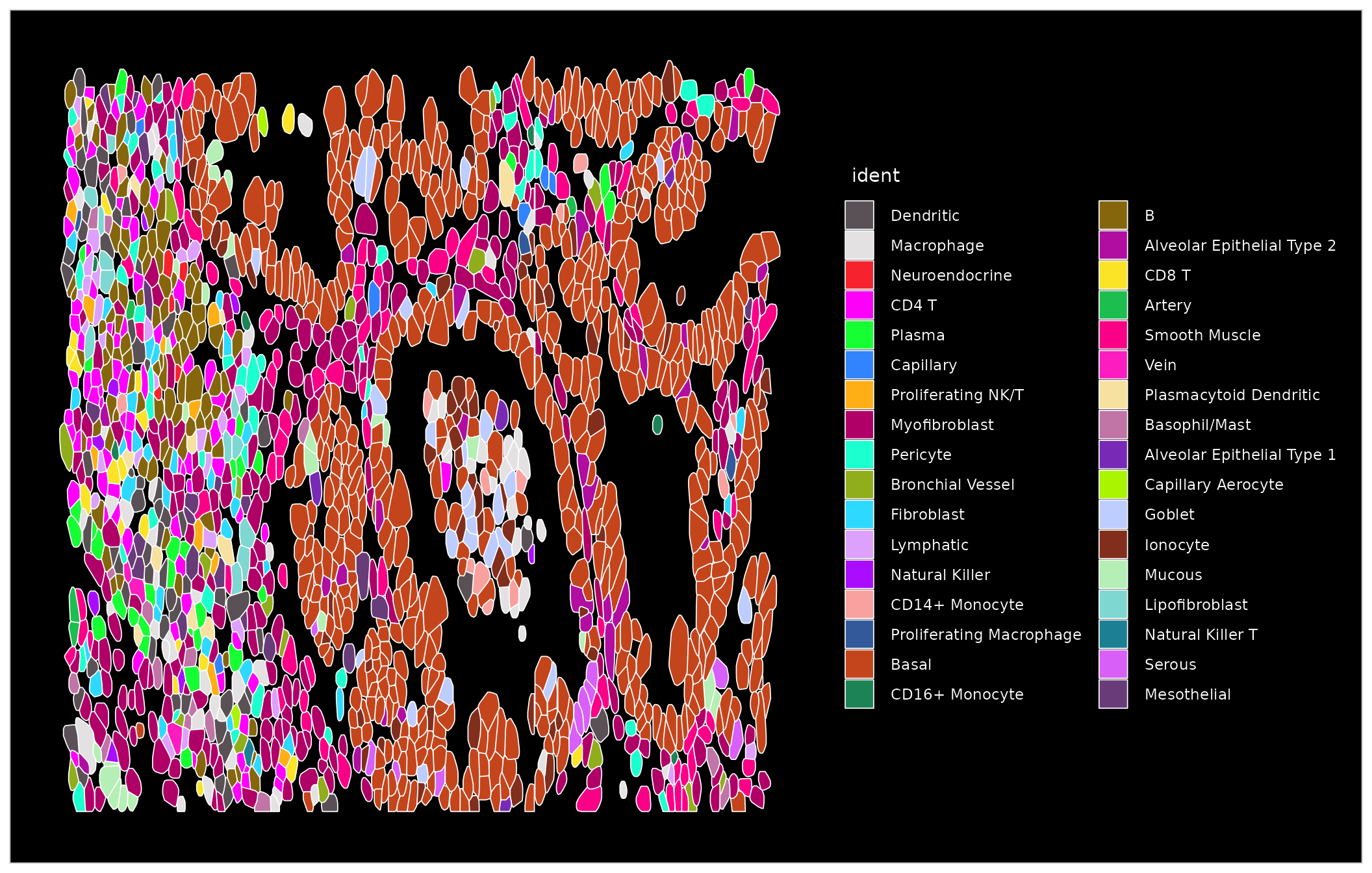
我们使用 Crop() 函数放大一个富含基底的区域。放大后,我们可以在所有可视化中可视化单个细胞边界。
basal.crop <- Crop(nano.obj[["lung5.rep1"]], x = c(159500, 164000), y = c(8700, 10500))
nano.obj[["zoom1"]] <- basal.crop
DefaultBoundary(nano.obj[["zoom1"]]) <- "segmentation"
ImageDimPlot(nano.obj, fov = "zoom1", cols = "polychrome", coord.fixed = FALSE)
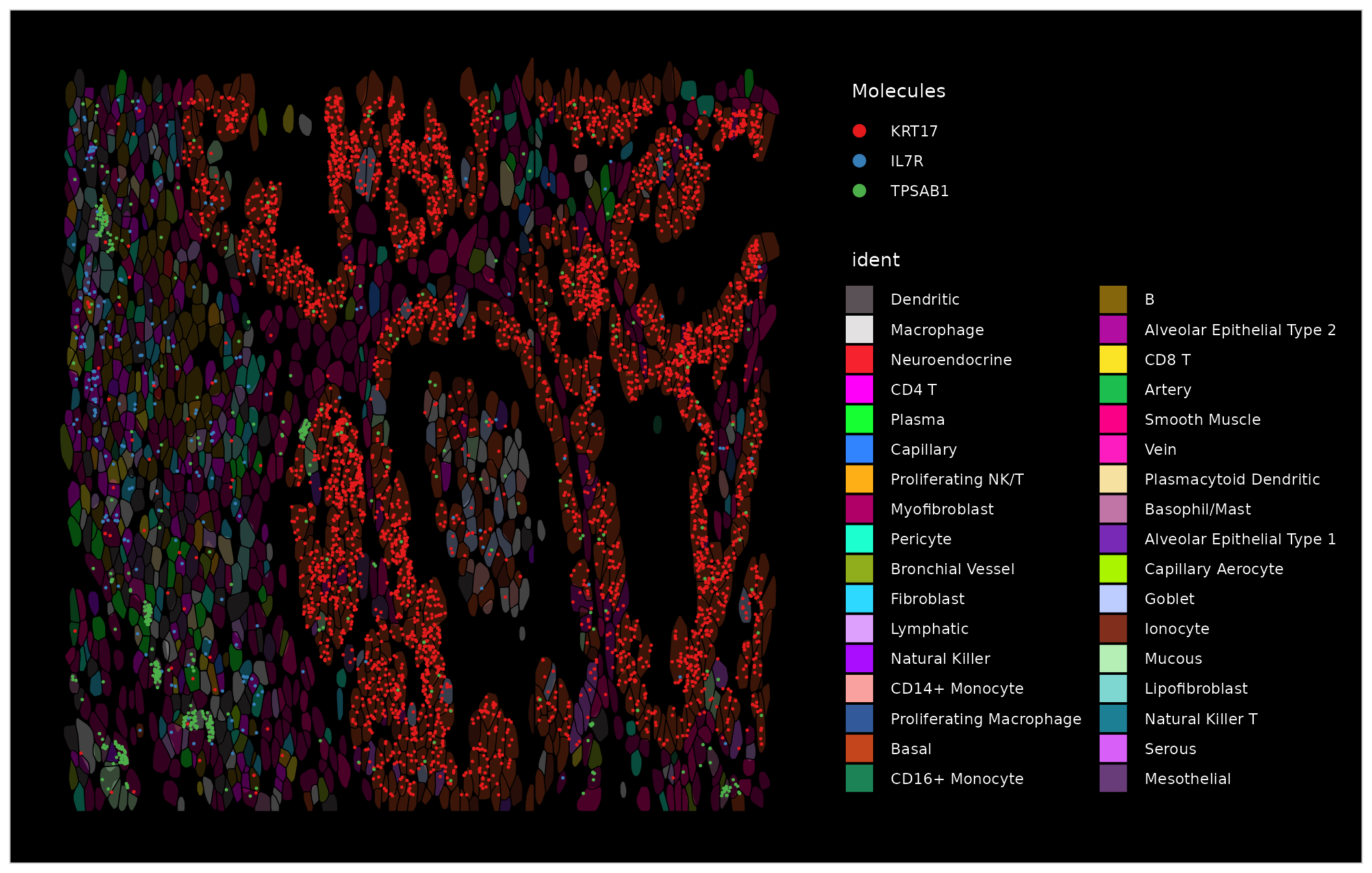
# note the clouds of TPSAB1 molecules denoting mast cells
ImageDimPlot(nano.obj, fov = "zoom1", cols = "polychrome", alpha = 0.3, molecules = c("KRT17", "IL7R",
"TPSAB1"), mols.size = 0.3, nmols = 20000, border.color = "black", coord.fixed = FALSE)








 苏公网安备32011502012024号
苏公网安备32011502012024号