引言
本文介绍了如何在Seurat软件中将查询数据集与经过注释的参考数据集进行匹配。以一个实例来说,我们把10X Genomics公司早期发布的一个包含2700个外周血单核细胞(PBMC)的单细胞RNA测序(scRNA-seq)数据集,与我们最近创建的一个使用228种抗体测量的、包含162,000个PBMC的CITE-seq参考数据集进行匹配。这个例子用来说明,在参考数据集的帮助下进行的有监督分析,是如何帮助我们识别那些仅通过无监督分析难以发现的细胞状态。在另一个例子中,我们展示了如何将来自不同个体的人类骨髓细胞(Human BMNC)的人类细胞图谱(Human Cell Atlas)数据集,有序地映射到一个统一的参考框架上。
我们之前利用参考映射的方法来标注查询数据集中的细胞标签。在Seurat v4版本中,大幅提高了执行集成任务,包括参考映射的速度和内存效率,并且还新增了将查询细胞投影到之前计算好的UMAP(Uniform Manifold Approximation and Projection,均匀流形近似和投影)可视化界面的功能。
内容
在本示例中,我们将展示如何利用一个已经建立的参考数据集来解读单细胞RNA测序(scRNA-seq)查询:
- 根据参考数据集定义的细胞状态集,对每个查询细胞进行标注。
- 将每个查询细胞投影到之前计算完成的UMAP可视化界面上。
- 估算在CITE-seq参考数据集中测量到的表面蛋白的预测水平。
要运行本示例,请确保安装了Seurat v4,该软件可在CRAN上下载。同时,您还需要安装SeuratDisk包。
library(Seurat)
library(ggplot2)
library(patchwork)
options(SeuratData.repo.use = "http://seurat.nygenome.org")
Example 1:绘制人类外周血细胞图谱
Data
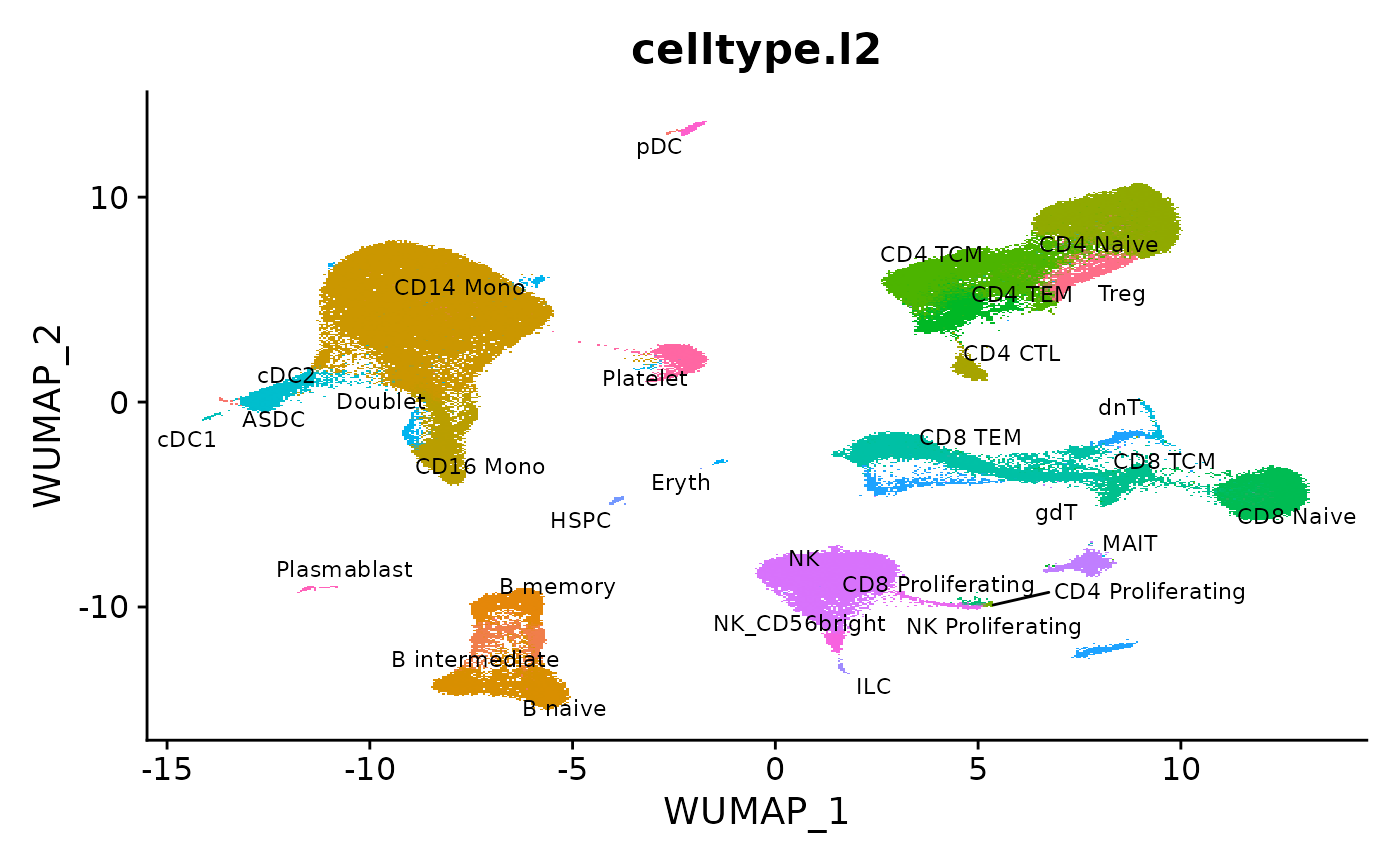
我们从最近的论文 中加载 reference,并可视化预先计算的 UMAP。
中加载 reference,并可视化预先计算的 UMAP。
reference <- readRDS("/brahms/hartmana/vignette_data/pbmc_multimodal_2023.rds")
DimPlot(object = reference, reduction = "wnn.umap", group.by = "celltype.l2", label = TRUE, label.size = 3, repel = TRUE) + NoLegend()
Mapping
为了演示与此多模式参考的映射,我们将使用由 10x Genomics 生成并可通过 SeuratData 获取的 2,700 个 PBMC 数据集。
library(SeuratData)
InstallData('pbmc3k')
pbmc3k <- LoadData('pbmc3k')
pbmc3k <- UpdateSeuratObject(pbmc3k)
参考集是通过 SCTransform() 函数进行标准化处理的,因此我们同样采用这一方法来对查询集进行标准化处理。
pbmc3k <- SCTransform(pbmc3k, verbose = FALSE)
接下来,我们在参考集与查询集之间确立对应关系。根据论文中的描述,本例中我们采用了预先计算的监督主成分分析(Supervised PCA,简称spca)变换。我们建议对CITE-seq数据集采用监督主成分分析方法,并将在本指南的下一个部分展示如何执行这一变换。当然,您也可以选择使用传统的主成分分析(PCA)变换。
anchors <- FindTransferAnchors(
reference = reference,
query = pbmc3k,
normalization.method = "SCT",
reference.reduction = "spca",
dims = 1:50
)
接下来,我们将细胞类型的标签和蛋白质信息从参考集转移到查询集。同时,我们还把查询集的数据映射到参考集的UMAP(均匀流形逼近和投影)结构上。
pbmc3k <- MapQuery(
anchorset = anchors,
query = pbmc3k,
reference = reference,
refdata = list(
celltype.l1 = "celltype.l1",
celltype.l2 = "celltype.l2",
predicted_ADT = "ADT"
),
reference.reduction = "spca",
reduction.model = "wnn.umap"
)
MapQuery() 函数封装了三个子函数:TransferData()、IntegrateEmbeddings() 和 ProjectUMAP()。TransferData() 函数的作用是传递细胞类型的标签并估算 ADT(Ambient Dolly Technology)的数值。而 IntegrateEmbeddings() 和 ProjectUMAP() 函数则用于将查询数据集映射到参考数据集的 UMAP(均匀流形逼近和投影)结构上。以下是使用这些中间函数完成相同操作的代码示例:
pbmc3k <- TransferData(
anchorset = anchors,
reference = reference,
query = pbmc3k,
refdata = list(
celltype.l1 = "celltype.l1",
celltype.l2 = "celltype.l2",
predicted_ADT = "ADT")
)
pbmc3k <- IntegrateEmbeddings(
anchorset = anchors,
reference = reference,
query = pbmc3k,
new.reduction.name = "ref.spca"
)
pbmc3k <- ProjectUMAP(
query = pbmc3k,
query.reduction = "ref.spca",
reference = reference,
reference.reduction = "spca",
reduction.model = "wnn.umap"
)
探索映射结果
目前,我们能够对2700个查询细胞进行可视化展示。这些细胞已经被映射到由参考数据集定义的UMAP(均匀流形逼近和投影)视图上,并且每个细胞都获得了两个不同详细程度(第一级和第二级)的注释信息。
p1 = DimPlot(pbmc3k, reduction = "ref.umap", group.by = "predicted.celltype.l1", label = TRUE, label.size = 3, repel = TRUE) + NoLegend()
p2 = DimPlot(pbmc3k, reduction = "ref.umap", group.by = "predicted.celltype.l2", label = TRUE, label.size = 3 ,repel = TRUE) + NoLegend()
p1 + p2
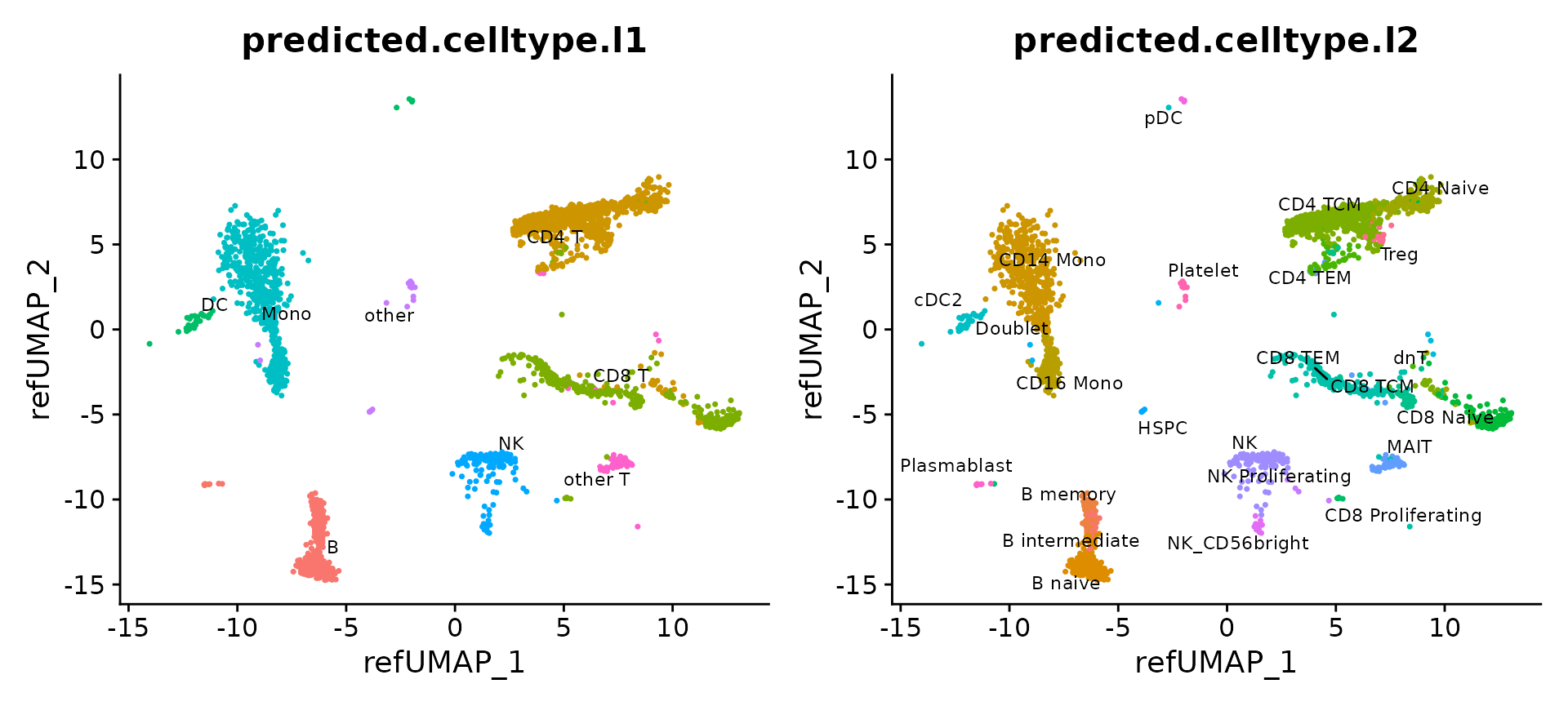
通过参考映射数据集,我们能够辨识出在对查询数据集进行无监督分析时难以区分的细胞类型。例如,这包括了浆细胞样树突状细胞(pDC)、造血干细胞和祖细胞(HSPC)、调节性T细胞(Treg)、CD8 初始T细胞、CD56+ 自然杀伤(NK)细胞、记忆B细胞和初始B细胞以及浆细胞。对于每个预测的细胞类型,系统都会给出一个0到1范围内的评分。
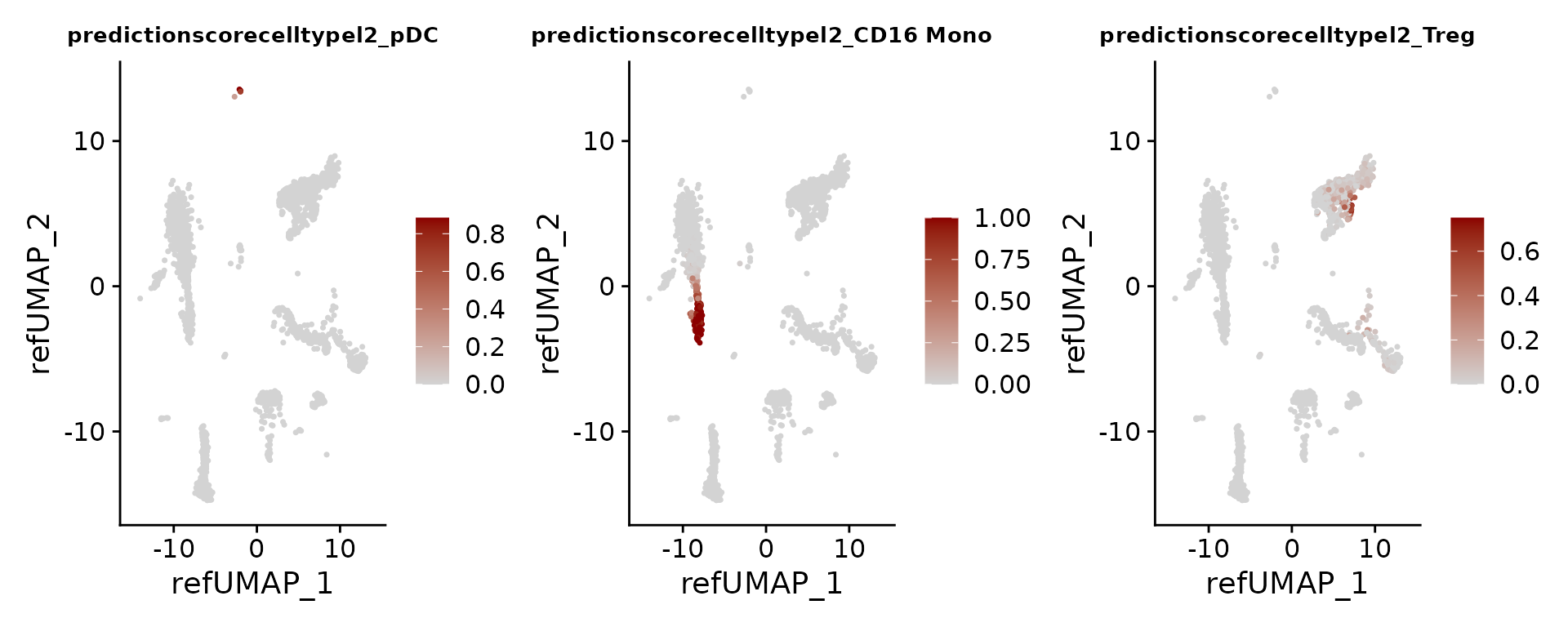
FeaturePlot(pbmc3k, features = c("pDC", "CD16 Mono", "Treg"), reduction = "ref.umap", cols = c("lightgrey", "darkred"), ncol = 3) & theme(plot.title = element_text(size = 10))
library(ggplot2)
plot <- FeaturePlot(pbmc3k, features = "CD16 Mono", reduction = "ref.umap", cols = c("lightgrey", "darkred")) + ggtitle("CD16 Mono") + theme(plot.title = element_text(hjust = 0.5, size = 30)) + labs(color = "Prediction Score") + xlab("UMAP 1") + ylab("UMAP 2") +
theme(axis.title = element_text(size = 18), legend.text = element_text(size = 18), legend.title = element_text(size = 25))
ggsave(filename = "../output/images/multimodal_reference_mapping.jpg", height = 7, width = 12, plot = plot, quality = 50)
我们可以通过检查特定标记基因的表达模式来核实我们的预测结果。举例来说,已有研究指出CLEC4C和LIR4是浆细胞样树突状细胞(pDC)的标志性基因,这与我们的预测相符。同样,如果我们通过差异表达分析来筛选调节性T细胞(Treg)的标记,我们能够识别出一组标准标记基因,包括RTKN2、CTLA4、FOXP3和IL2RA。
Idents(pbmc3k) <- 'predicted.celltype.l2'
VlnPlot(pbmc3k, features = c("CLEC4C", "LILRA4"), sort = TRUE) + NoLegend()

treg_markers <- FindMarkers(pbmc3k, ident.1 = "Treg", only.pos = TRUE, logfc.threshold = 0.1)
print(head(treg_markers))
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## AC004854.4 4.795409e-26 7.800900 0.042 0.000 6.028789e-22
## RP11-297N6.4 4.795409e-26 7.800900 0.042 0.000 6.028789e-22
## RTKN2 4.795409e-26 7.800900 0.042 0.000 6.028789e-22
## CTD-2020K17.4 4.795409e-26 7.800900 0.042 0.000 6.028789e-22
## RP11-1399P15.1 2.782841e-18 4.172869 0.292 0.021 3.498588e-14
## IL2RA 1.808612e-14 4.137935 0.208 0.014 2.273787e-10
最终,我们能够展示根据 CITE-seq 参考数据推断出的表面蛋白表达水平的可视化结果。
DefaultAssay(pbmc3k) <- 'predicted_ADT'
# see a list of proteins: rownames(pbmc3k)
FeaturePlot(pbmc3k, features = c("CD3-1", "CD45RA", "IgD"), reduction = "ref.umap", cols = c("lightgrey", "darkgreen"), ncol = 3)

计算新的 UMAP 可视化
在之前的案例中,我们将查询细胞映射到基于参考数据集生成的 UMAP 结构后进行了可视化展示。维持一致的可视化方式有助于我们解读新的数据集。然而,如果查询数据集中包含了在参考数据集中未出现的细胞状态,这些状态将会映射到参考数据集中最为相似的细胞类型上。这是 UMAP 软件包所预期的工作方式,但有时也可能因此忽视了查询数据集中存在的、可能具有研究价值的新细胞类型。
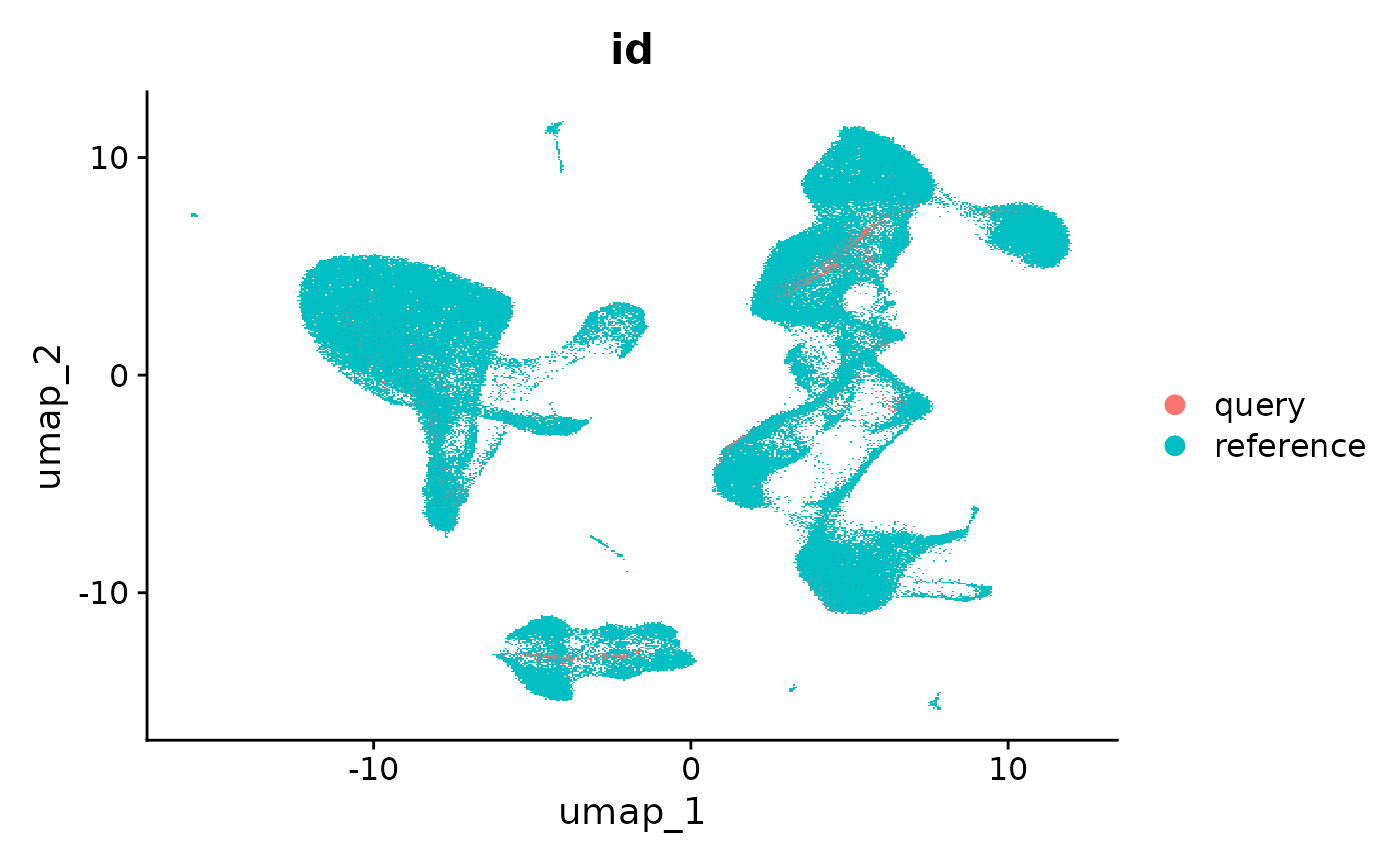
在我们的论文中,我们对一个包含发育中和已分化的中性粒细胞的查询数据集进行了映射,这些细胞在我们的参考数据集中并未包含。我们发现,在将参考数据集和查询数据集合并后,重新计算一个新的 UMAP(称为“从头可视化”)有助于识别这些细胞群体,这一点在补充图 8 中有展示。在“从头可视化”中,查询数据集中的独特细胞状态仍然保持独立。在这个例子中,2700个外周血单核细胞(PBMC)并没有包含独特的细胞状态,但我们将展示如何计算这种可视化。
我们想强调的是,如果用户尝试映射的数据集样本不是 PBMC,或者包含了参考数据集中未出现细胞类型,那么进行一次“从头可视化”是理解和分析他们数据集的一个重要步骤。
reference <- DietSeurat(reference, counts = FALSE, dimreducs = "spca")
pbmc3k <- DietSeurat(pbmc3k, counts = FALSE, dimreducs = "ref.spca")
#merge reference and query
reference$id <- 'reference'
pbmc3k$id <- 'query'
refquery <- merge(reference, pbmc3k)
refquery[["spca"]] <- merge(reference[["spca"]], pbmc3k[["ref.spca"]])
refquery <- RunUMAP(refquery, reduction = 'spca', dims = 1:50)
DimPlot(refquery, group.by = 'id', shuffle = TRUE)








 苏公网安备32011502012024号
苏公网安备32011502012024号