细胞级别的片段分布
CoveragePlot() 函数通常用于计算基因组区域内不同细胞群体的信号总和,但有时候,也需要单独查看单个细胞在基因组区域内的序列化片段频率,而不是将它们聚合起来。这时,TilePlot() 函数就能派上用场。
tile_plot <- TilePlot(
object = pbmc,
region = roi,
idents = c("CD4 Memory", "CD8 Effector")
)
tile_plot
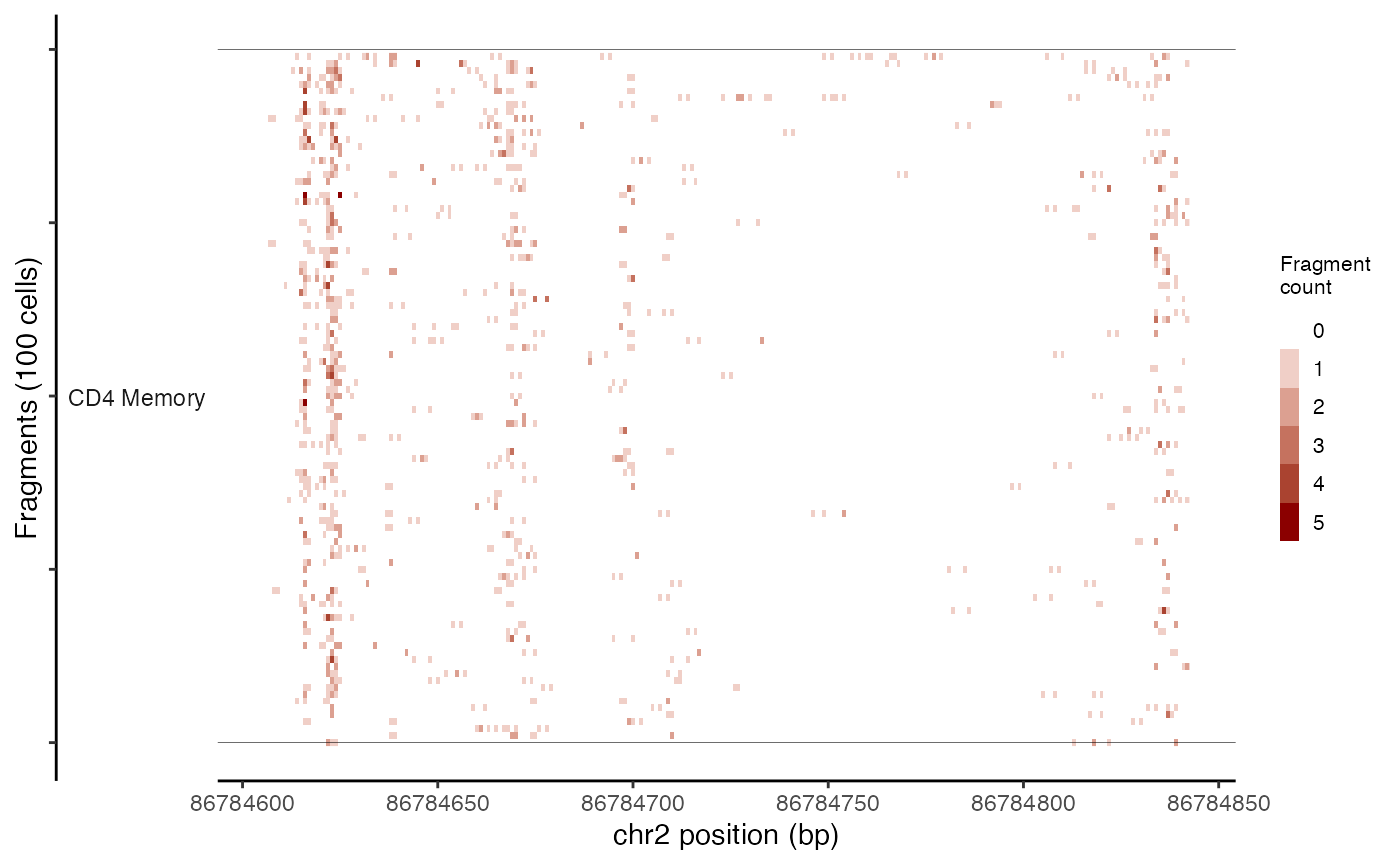
默认情况下,系统会自动为每个组挑选出基因组区域内总片段数最多的前100个细胞。接着,基因组区域会被划分成多个小区域,对每个细胞在这些小区域中的片段总数进行计数,并将这些计数结果以热图的形式展示出来。
在基因组轨迹图上添加额外的数据
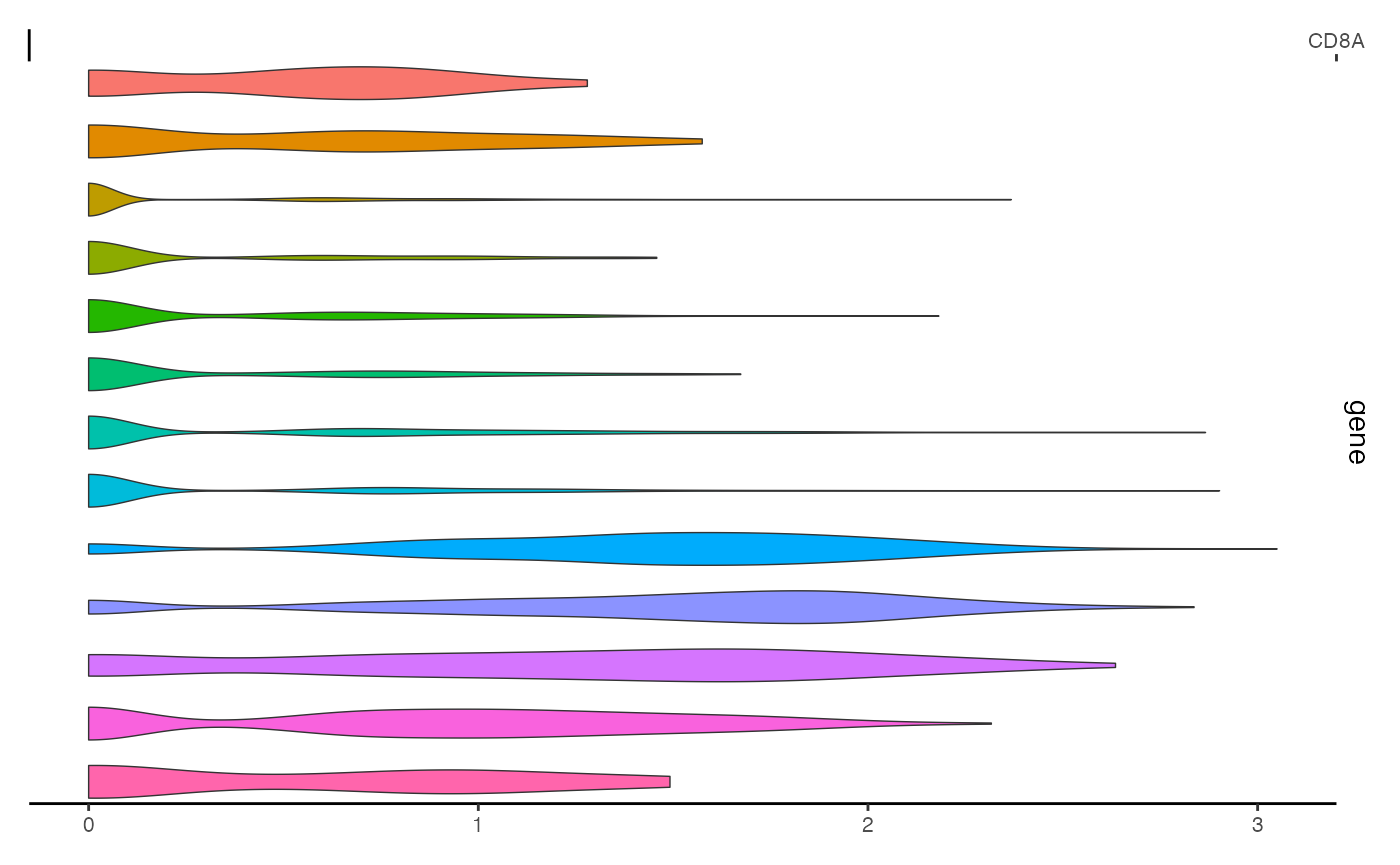
多模态单细胞数据集能够为每个细胞提供多种实验测量结果。目前,已经有一些方法能够同时测量单细胞的染色质数据(比如染色质的可及性)以及同一细胞的其他数据,比如基因表达或线粒体基因型。在这种情况下,将这些多模态数据整合到一张图表中进行可视化,往往能提供更多的信息。这可以通过ExpressionPlot()函数来实现,它的功能类似于Seurat中的VlnPlot()函数,但专门设计用来与CoveragePlot()生成的基因组轨迹图相结合。
expr_plot <- ExpressionPlot(
object = pbmc,
features = "CD8A",
assay = "RNA"
)
expr_plot
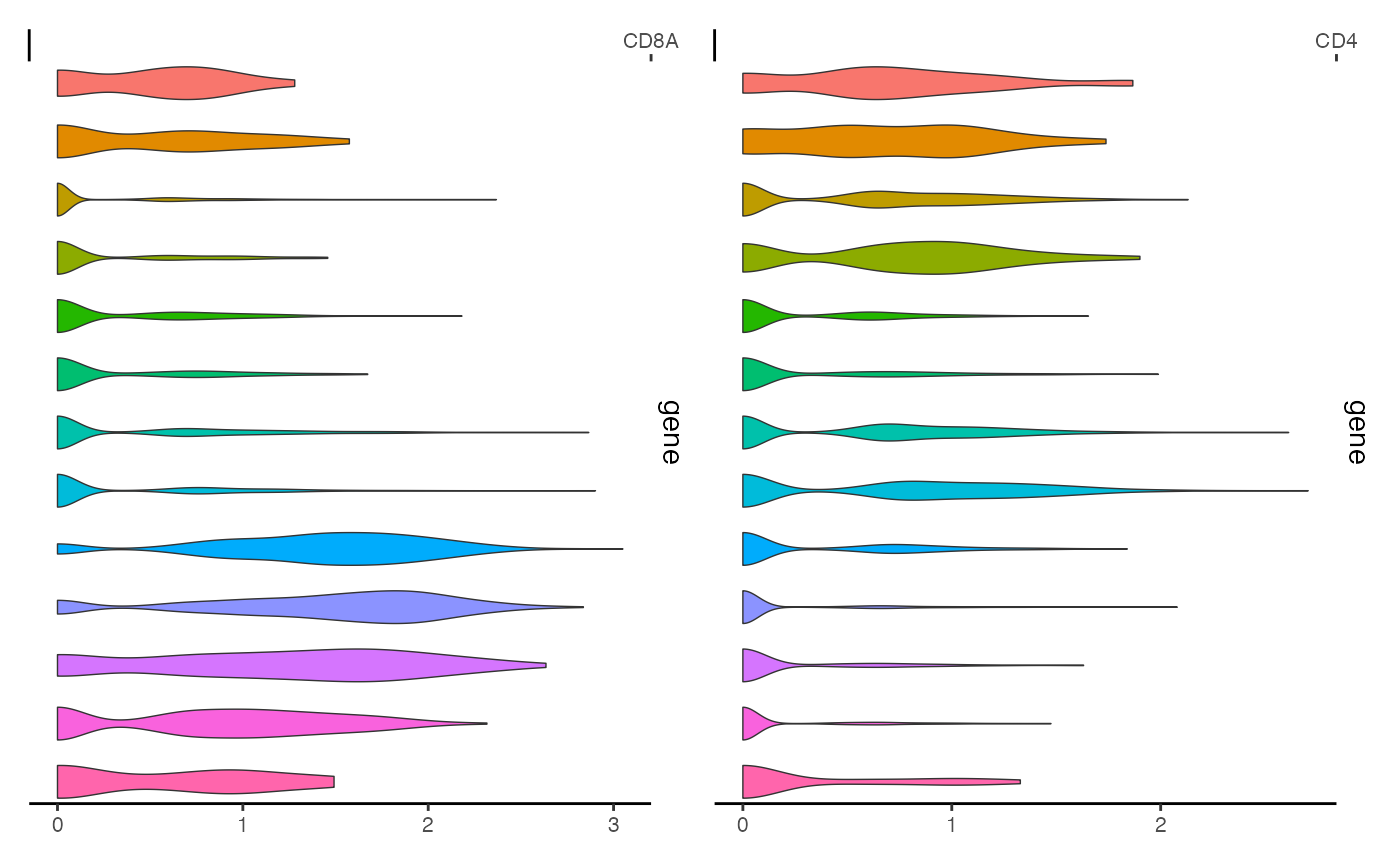
可以通过传递基因名称列表来同时为多个基因创建类似的图
ExpressionPlot(
object = pbmc,
features = c("CD8A", "CD4"),
assay = "RNA"
)
合并基因组轨迹图
之前已经介绍了如何创建单独的轨迹和图表,现在可以将它们合并成一个基因组区域的总图。使用 CombineTracks() 函数可以很方便地实现这种合并。
CombineTracks(
plotlist = list(cov_plot, tile_plot, peak_plot, gene_plot, link_plot),
expression.plot = expr_plot,
heights = c(10, 6, 1, 2, 3),
widths = c(10, 1)
)
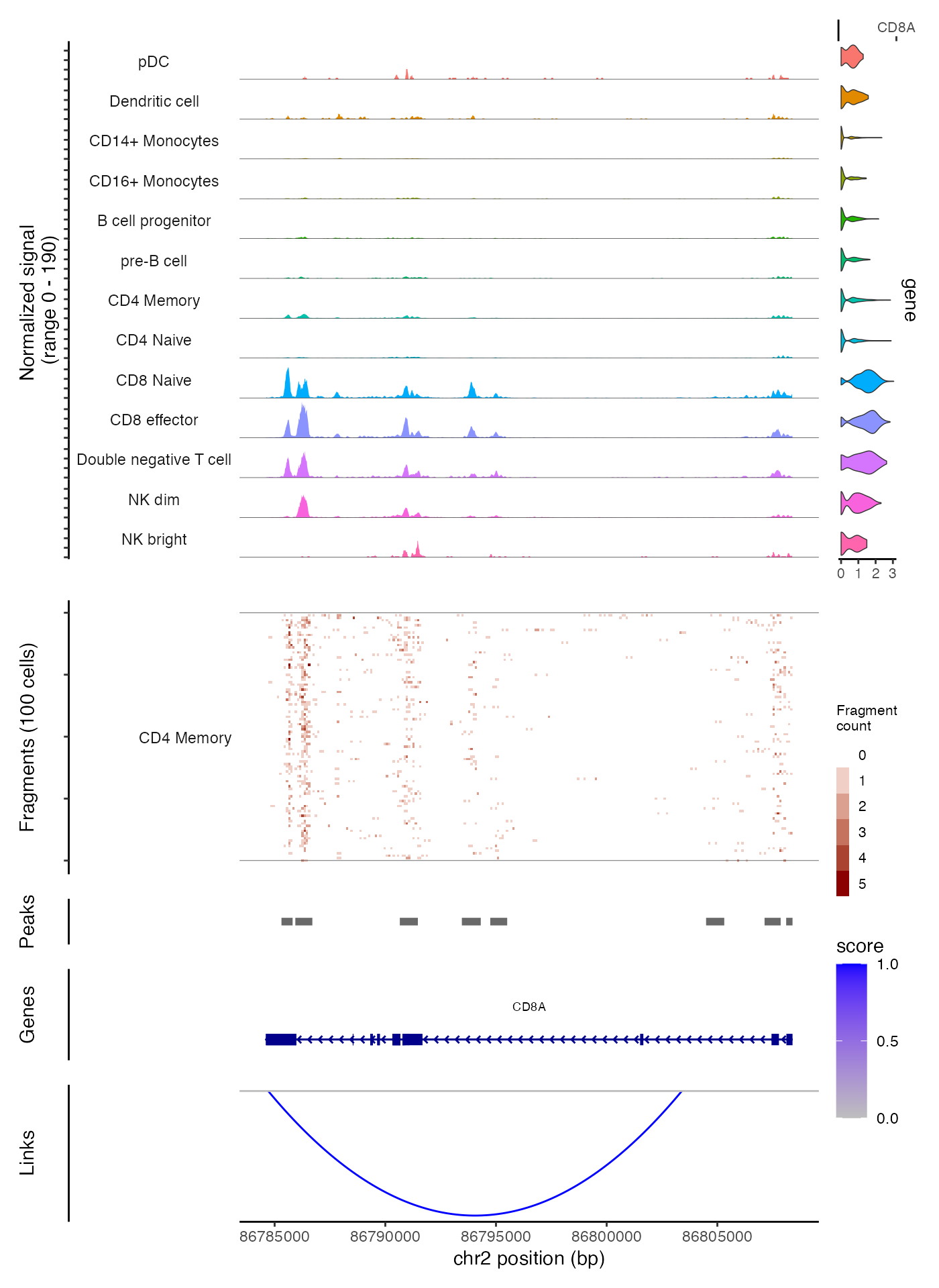
"heights" 和 "widths" 参数用于调整各个轨迹图的相对高度和宽度,调整的顺序依据它们在 plotlist 中的排列。CombineTracks() 函数确保这些轨迹在垂直和水平方向上整齐对齐,并将描述基因组位置的 x 轴标签移到组合轨迹图的底部。
批量生成多个轨迹图
之前介绍了如何分别创建基因组轨迹图面板以及如何将它们合并。这种方法虽然可以更细致地控制每个面板的构建和合并过程,但步骤较多。为了简化操作,在 CoveragePlot() 函数中提供了一种自动生成和合并不同面板的功能,通过 annotation、peaks、tile 和 features 参数即可实现。这样,可以通过一次函数调用,快速生成类似于前面展示的图表:
CoveragePlot(
object = pbmc,
region = roi,
features = "CD8A",
annotation = TRUE,
peaks = TRUE,
tile = TRUE,
links = TRUE
)
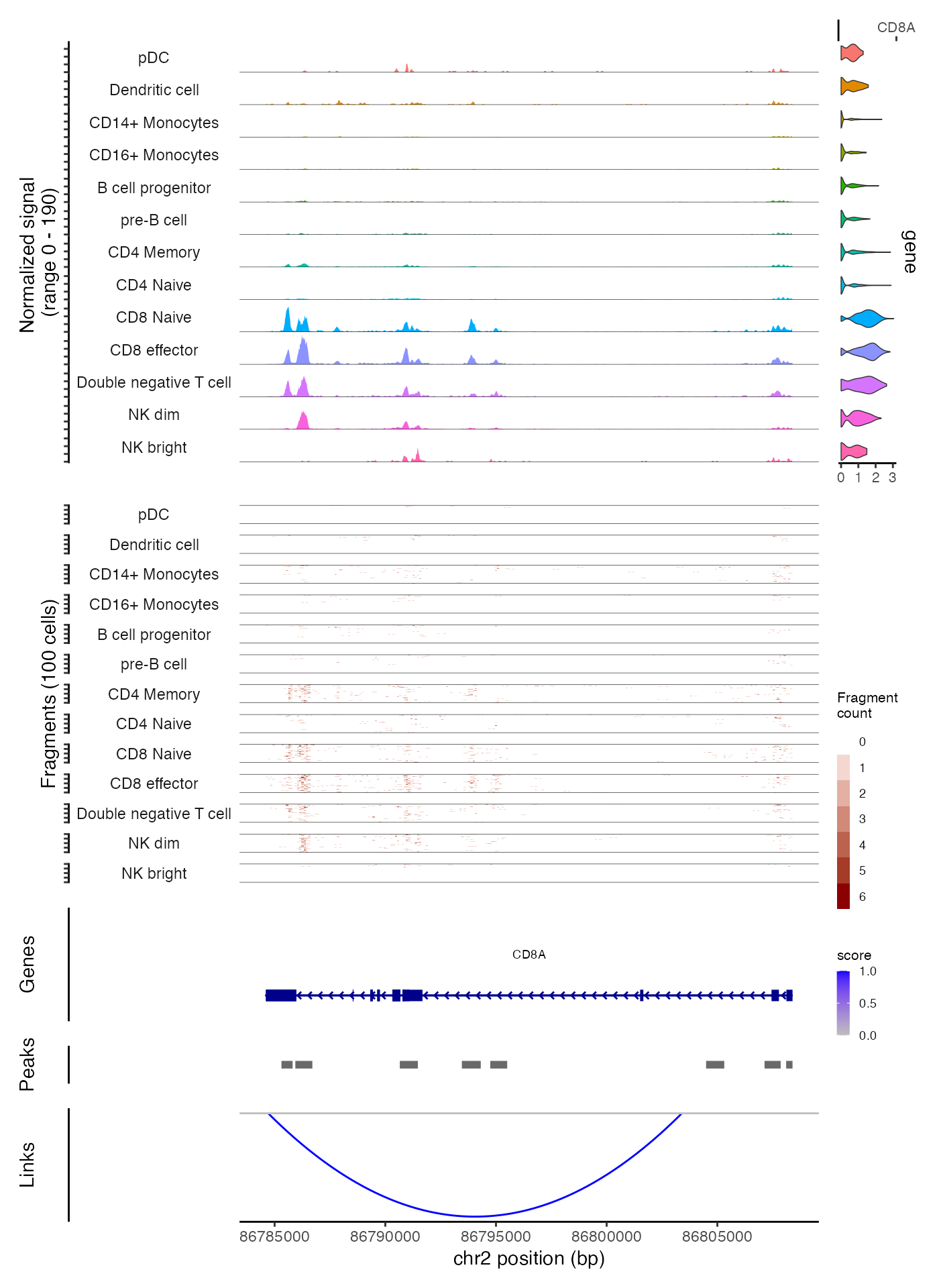
在这个示例中,为覆盖轨迹中展示的每个细胞群生成了瓦片图,而在之前的示例中,能够生成一个图表,它不仅展示了所有细胞群的总体覆盖情况,还特别展示了CD4记忆细胞和CD8效应细胞的瓦片图。当分别创建每个轨迹时,可以进行更高程度的个性化定制。








 苏公网安备32011502012024号
苏公网安备32011502012024号