数据和代码来源于论文
Using genomics, morphometrics, and environmental niche modeling to test the validity of a narrow-range endemic snail, Patera nantahala (Gastropoda, Polygyridae)
https://github.com/NathanWhelan/Patera/blob/main/niche-modeling_Patera_FINAL.R
https://figshare.com/articles/dataset/Using_genomics_morphometrics_and_environmental_niche_modeling_to_test_the_validity_of_a_narrow-range_endemic_snail_Patera_nantahala_GastropodaPolygyridae/19638642
如果要做这个分析的话需要准备的数据是
- 两个不同物种的经纬度数据
- 环境数据(这个环境数据可以利用geodata::worldclim_global下载)
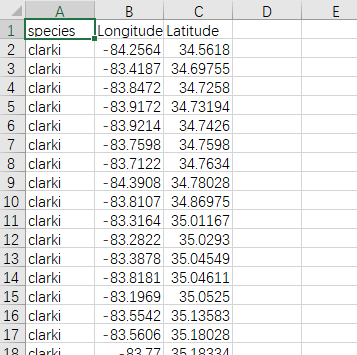
经纬度数据的格式
代码
library(tidyverse)
library(ENMTools)
sp1 <- enmtools.species(species.name = "sp1",
presence.points = read.csv("2024.data/20240612/clarki.csv"))
sp2 <- enmtools.species(species.name = "sp2",
presence.points = read.csv("2024.data/20240612/nantahala.csv"))
tif.path<-paste0("2024.data/20240612/climate/wc2.1_5m/wc2.1_5m_bio_",1:19,".tif")
tif.path
wclim<-terra::rast(tif.path)
## 环境数据是全球范围的数据,
## 裁剪成一定范围,根据物种的分布来裁剪
wclim.crop <- crop(wclim, extent(-85, -80, 34, 37))
reps <- 10
## 重复次数设置100以上,这里只是为了练习
back <- 10000
idtest_glm <- identity.test(
sp1,
sp2,
wclim.crop,
type = 'glm',
f = NULL,
nreps = reps,
nback = back,
low.memory = FALSE,
bg.source="env",
rep.dir = "bioclimate_glm",
verbose = FALSE,
clamp = TRUE,
)
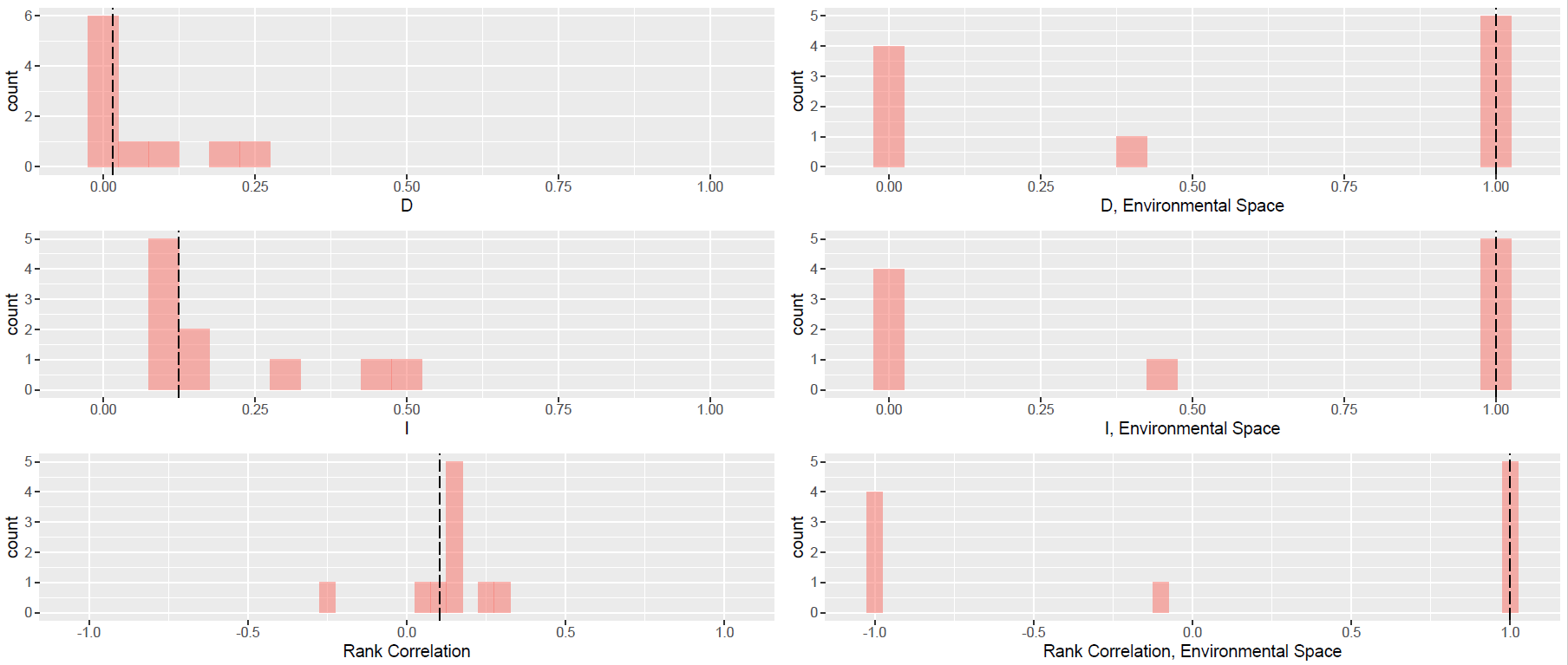
idtest_glm
idtest_glm$p.values
idtest_glm$description
idtest_glm$d.plot
idtest_glm$i.plot
关于结果的描述应该按照这个论文来写就行
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0290044
所有的测试代码
library(tidyverse)
library(ENMTools)
# wclim<-geodata::worldclim_global(var="bio", res=5,
# path = "2024.data/20240612/")
tif.path<-paste0("2024.data/20240612/climate/wc2.1_5m/wc2.1_5m_bio_",1:19,".tif")
tif.path
wclim<-terra::rast(tif.path)
plot(wclim[[1]])
wclim.crop <- crop(wclim, extent(-85, -80, 34, 37))
plot(wclim.crop[[1]])
x<-read_csv("2024.data/20240612/clarki-nantahala_1KM-thinned_rarefied_points.csv",
col_names = FALSE)
x
colnames(x)[1:3]<-c("species","Longitude","Latitude")
x %>% pull(Longitude) %>% range()
x %>% pull(Latitude) %>% range()
world.dat<-map_data("world")
ggplot() +
geom_polygon(data=world.dat,aes(x=long,y=lat,group=group),
fill="#dedede")+
geom_point(data = x,aes(x=Longitude,y=Latitude))
x %>%
pull(species) %>%
table()
x %>%
filter(species=="clarki") %>%
write_csv("2024.data/20240612/clarki.csv")
x %>%
filter(species=="nantahala") %>%
write_csv("2024.data/20240612/nantahala.csv")
sp1 <- enmtools.species(species.name = "sp1",
presence.points = read.csv("2024.data/20240612/clarki.csv"))
sp2 <- enmtools.species(species.name = "sp2",
presence.points = read.csv("2024.data/20240612/nantahala.csv"))
tif.path<-paste0("2024.data/20240612/climate/wc2.1_5m/wc2.1_5m_bio_",1:19,".tif")
tif.path
wclim<-terra::rast(tif.path)
wclim.crop <- crop(wclim, extent(-85, -80, 34, 37))
reps <- 10
back <- 10000
idtest_glm <- identity.test(
sp1,
sp2,
wclim.crop,
type = 'glm',
f = NULL,
nreps = reps,
nback = back,
low.memory = FALSE,
bg.source="env",
rep.dir = "bioclimate_glm",
verbose = FALSE,
clamp = TRUE,
)
idtest_glm$p.values
idtest_glm$description
idtest_glm$d.plot
idtest_glm$i.plot
idtest_glm
欢迎大家关注我的公众号
小明的数据分析笔记本
小明的数据分析笔记本 公众号 主要分享:1、R语言和python做数据分析和数据可视化的简单小例子;2、园艺植物相关转录组学、基因组学、群体遗传学文献阅读笔记;3、生物信息学入门学习资料及自己的学习笔记!





 苏公网安备32011502012024号
苏公网安备32011502012024号