引言
在本指南中,将介绍如何利用Cicero工具和单细胞ATAC-seq数据来识别共可接近网络。
为了在Seurat(Signac工具使用的格式)和CellDataSet(Cicero工具使用的格式)之间轻松转换数据,将利用GitHub上的SeuratWrappers包提供的转换功能。
数据加载
将采用Satpathy和Granja等人在2019年发表在《Nature Biotechnology》上的研究成果,使用他们发布的包含人类CD34+造血干细胞和祖细胞的单细胞ATAC-seq数据集。这些经过处理的数据可以在NCBI GEO数据库中找到链接:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129785
首先,会加载他们的数据集,并使用Signac工具进行一些常规的预处理工作。
library(Signac)
library(Seurat)
library(SeuratWrappers)
library(ggplot2)
library(patchwork)
# load the object created in the Monocle 3 vignette
bone <- readRDS("cd34.rds")
构建 Cicero 模型
Cicero 工具能够识别共可接近网络(CCANs)。Cicero 的开发团队已经创建了一个特别分支,该分支能够与 Monocle 3 的 CellDataSet 对象协同工作。我们首先需要确保已经安装了这个分支,然后我们将整个骨髓数据集的 Seurat 对象转换成 CellDataSet 格式。
# Install Cicero
if (!requireNamespace("remotes", quietly = TRUE))
install.packages("remotes")
remotes::install_github("cole-trapnell-lab/cicero-release", ref = "monocle3")
library(cicero)
# convert to CellDataSet format and make the cicero object
bone.cds <- as.cell_data_set(x = bone)
bone.cicero <- make_cicero_cds(bone.cds, reduced_coordinates = reducedDims(bone.cds)$UMAP)
探索 Cicero 连接
为了节省时间,将在这里仅用一个染色体来演示如何运行 Cicero,但同样的流程也可以用来在整个基因组中寻找共可接近网络(CCANs)。
接下来,将展示运行 Cicero 的基础步骤。这个流程包含多个环节,每个环节的参数都可以根据您的数据需求从默认设置中调整,以优化 Cicero 算法的性能。强烈推荐访问 Cicero 的官方网站、相关论文和文档,以获取更详尽的信息。
# get the chromosome sizes from the Seurat object
genome <- seqlengths(bone)
# use chromosome 1 to save some time
# omit this step to run on the whole genome
genome <- genome[1]
# convert chromosome sizes to a dataframe
genome.df <- data.frame("chr" = names(genome), "length" = genome)
# run cicero
conns <- run_cicero(bone.cicero, genomic_coords = genome.df, sample_num = 100)
head(conns)
## Peak1 Peak2 coaccess
## 1 chr1-100003337-100003837 chr1-99791719-99792219 0
## 2 chr1-100003337-100003837 chr1-99828699-99829199 0
## 3 chr1-100003337-100003837 chr1-99835542-99836042 0
## 4 chr1-100003337-100003837 chr1-99836217-99836717 0
## 5 chr1-100003337-100003837 chr1-99839576-99840076 0
## 6 chr1-100003337-100003837 chr1-99840640-99841140 0
识别 共可接近网络(CCANs)
既然已经计算出了每个峰值之间的共可接近性得分,现在可以利用 Cicero 工具中的 generate_ccans() 功能,将这些成对的联系整合成更广泛的共可接近网络。
ccans <- generate_ccans(conns)
head(ccans)
## Peak CCAN
## chr1-10009702-10010202 chr1-10009702-10010202 1
## chr1-100151188-100151688 chr1-100151188-100151688 2
## chr1-100164787-100165287 chr1-100164787-100165287 2
## chr1-100165566-100166066 chr1-100165566-100166066 2
## chr1-100202505-100203005 chr1-100202505-100203005 3
## chr1-100215491-100215991 chr1-100215491-100215991 3
将链接整合到 Seurat 对象
能够将 Cicero 识别出的共可接近链接整合进 Seurat 的 ChromatinAssay 对象中。通过 Signac 包中的 ConnectionsToLinks() 函数,可以将 Cicero 的输出转换成适合存储在 ChromatinAssay 对象链接槽的格式,然后利用 Links<- 赋值操作将这些链接添加到对象中。
links <- ConnectionsToLinks(conns = conns, ccans = ccans)
Links(bone) <- links
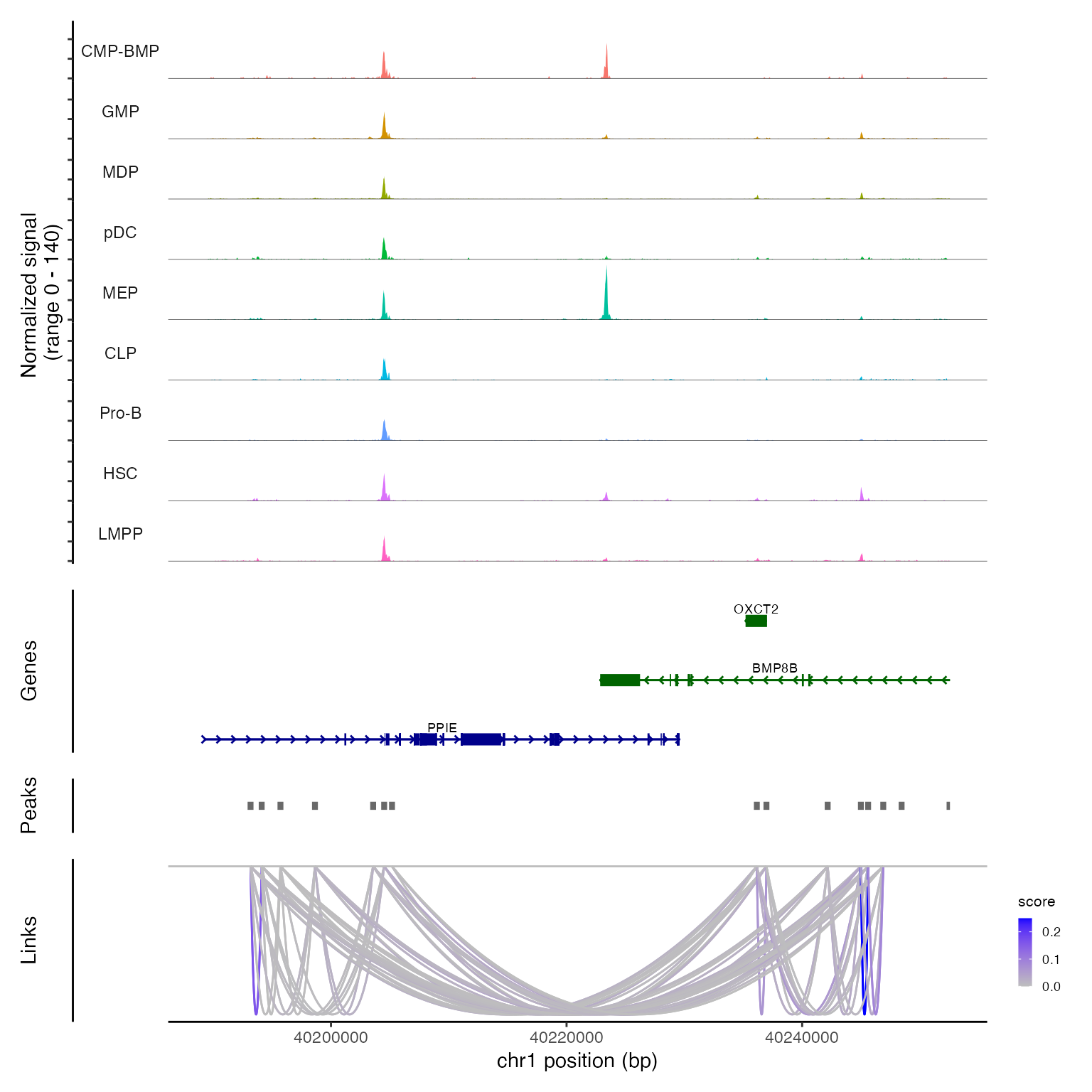
现在,可以通过对某个区域运行 CoveragePlot() 来可视化这些链接以及 DNA 可及性信息:
CoveragePlot(bone, region = "chr1-40189344-40252549")








 苏公网安备32011502012024号
苏公网安备32011502012024号