引言
本研究分析了两组包含DNA可及性和线粒体突变信息的数据集,数据来自结直肠癌(CRC)患者的样本。
为了展示如何联合分析线粒体DNA变异和染色质可及性,将通过一个实例来说明,该实例涉及对来自原发性结直肠腺癌的细胞进行分析。这些细胞样本中包含了恶性上皮细胞和肿瘤内部浸润的免疫细胞。
导入 DNA 可及性数据
首先导入单细胞染色质可及性测序(scATAC-seq)数据,并按照scATAC-seq数据的标准分析流程,构建一个Seurat对象。
library(Signac)
library(Seurat)
library(ggplot2)
library(patchwork)
library(EnsDb.Hsapiens.v75)
# load counts and metadata from cellranger-atac
counts <- Read10X_h5(filename = "CRC_v12-mtMask_mgatk.filtered_peak_bc_matrix.h5")
metadata <- read.csv(
file = "CRC_v12-mtMask_mgatk.singlecell.csv",
header = TRUE,
row.names = 1
)
# load gene annotations from Ensembl
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v75)
# change to UCSC style since the data was mapped to hg19
seqlevels(annotations) <- paste0('chr', seqlevels(annotations))
genome(annotations) <- "hg19"
# create object
crc_assay <- CreateChromatinAssay(
counts = counts,
sep = c(":", "-"),
annotation = annotations,
min.cells = 10,
fragments = 'CRC_v12-mtMask_mgatk.fragments.tsv.gz'
)
crc <- CreateSeuratObject(
counts = crc_assay,
assay = 'peaks',
meta.data = metadata
)
crc[["peaks"]]
## ChromatinAssay data with 81787 features for 3535 cells
## Variable features: 0
## Genome:
## Annotation present: TRUE
## Motifs present: FALSE
## Fragment files: 1
质控流程
能够对单细胞ATAC-seq(scATAC-seq)数据进行标准的质量评估,并依据这些评估指标剔除质量不佳的细胞。
# Augment QC metrics that were computed by cellranger-atac
crc$pct_reads_in_peaks <- crc$peak_region_fragments / crc$passed_filters * 100
crc$pct_reads_in_DNase <- crc$DNase_sensitive_region_fragments / crc$passed_filters * 100
crc$blacklist_ratio <- crc$blacklist_region_fragments / crc$peak_region_fragments
# compute TSS enrichment score and nucleosome banding pattern
crc <- TSSEnrichment(crc)
crc <- NucleosomeSignal(crc)
# visualize QC metrics for each cell
VlnPlot(crc, c("TSS.enrichment", "nCount_peaks", "nucleosome_signal", "pct_reads_in_peaks", "pct_reads_in_DNase", "blacklist_ratio"), pt.size = 0, ncol = 3)
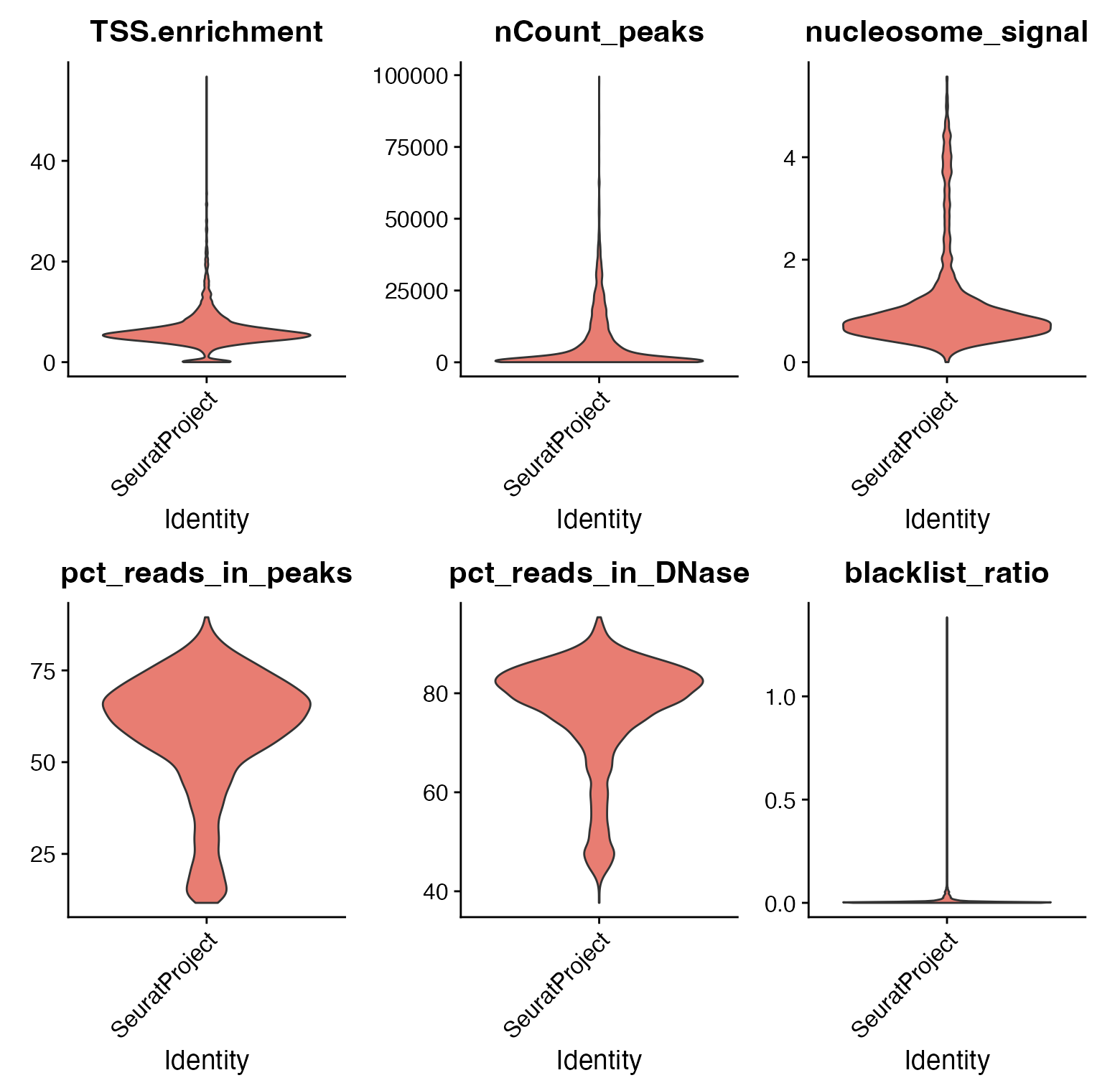
# remove low-quality cells
crc <- subset(
x = crc,
subset = nCount_peaks > 1000 &
nCount_peaks < 50000 &
pct_reads_in_DNase > 40 &
blacklist_ratio < 0.05 &
TSS.enrichment > 3 &
nucleosome_signal < 4
)
crc
## An object of class Seurat
## 81787 features across 1861 samples within 1 assay
## Active assay: peaks (81787 features, 0 variable features)
## 2 layers present: counts, data
线粒体变异数据导入
接下来,将导入这些细胞的线粒体DNA变异数据,这些数据是通过mgatk工具进行量化的。Signac包中的ReadMGATK()函数使得mgatk的输出能够直接以适合于Signac后续分析的格式读入R语言环境中。在此过程中,将这些数据加载进来,并将其作为一个新检测添加到Seurat对象中。
# load mgatk output
mito.data <- ReadMGATK(dir = "crc/")
# create an assay
mito <- CreateAssayObject(counts = mito.data$counts)
# Subset to cell present in the scATAC-seq assat
mito <- subset(mito, cells = Cells(crc))
# add assay and metadata to the seurat object
crc[["mito"]] <- mito
crc <- AddMetaData(crc, metadata = mito.data$depth[Cells(mito), ], col.name = "mtDNA_depth")
可以查看每个细胞的线粒体测序深度,并根据线粒体测序深度进一步对细胞进行子集化。
VlnPlot(crc, "mtDNA_depth", pt.size = 0.1) + scale_y_log10()
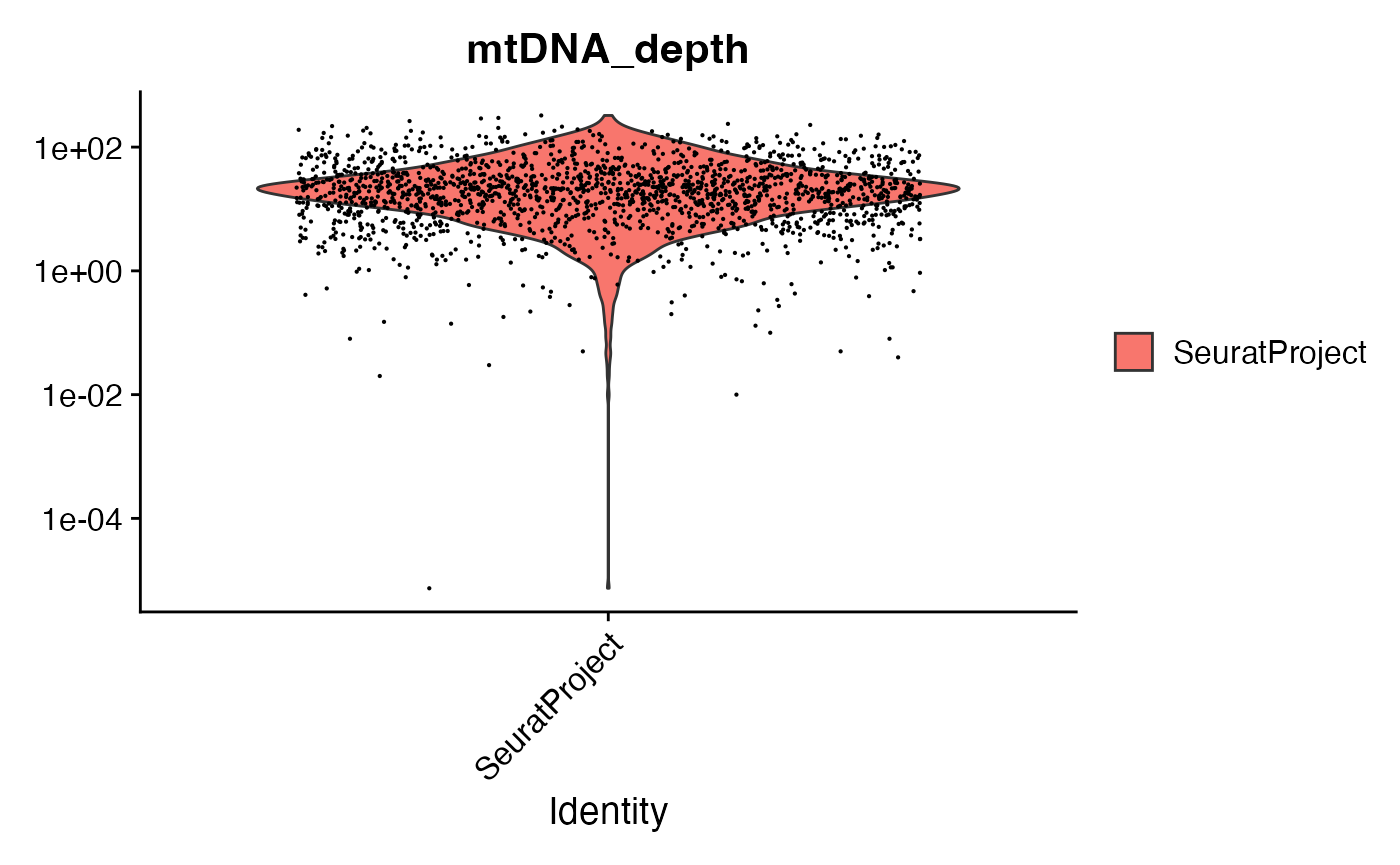
# filter cells based on mitochondrial depth
crc <- subset(crc, mtDNA_depth >= 10)
crc
## An object of class Seurat
## 214339 features across 1359 samples within 2 assays
## Active assay: peaks (81787 features, 0 variable features)
## 2 layers present: counts, data
## 1 other assay present: mito
降维和聚类
接下来,将利用单细胞ATAC-seq(scATAC-seq)数据进行常规的降维和聚类分析,目的是识别不同的细胞群组。
crc <- RunTFIDF(crc)
crc <- FindTopFeatures(crc, min.cutoff = 10)
crc <- RunSVD(crc)
crc <- RunUMAP(crc, reduction = "lsi", dims = 2:50)
crc <- FindNeighbors(crc, reduction = "lsi", dims = 2:50)
crc <- FindClusters(crc, resolution = 0.7, algorithm = 3)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 1359
## Number of edges: 63301
##
## Running smart local moving algorithm...
## Maximum modularity in 10 random starts: 0.7329
## Number of communities: 6
## Elapsed time: 0 seconds
DimPlot(crc, label = TRUE) + NoLegend()
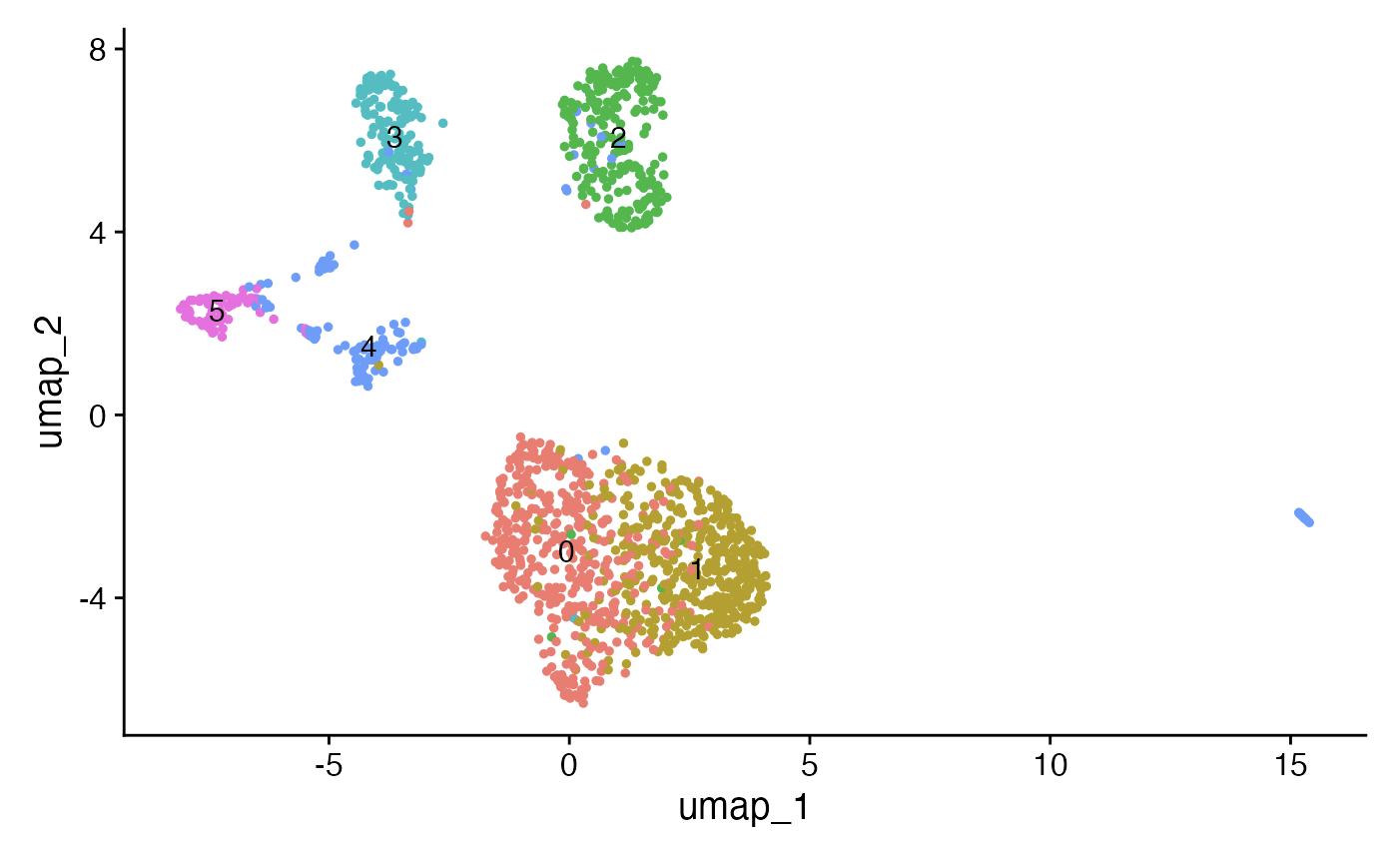
基因得分计算
为了更好地理解这些细胞群的特征并确定它们的细胞类型,将计算基因的活性得分,方法是将基因本体区域和启动子区域的DNA可及性进行累加。
# compute gene accessibility
gene.activities <- GeneActivity(crc)
# add to the Seurat object as a new assay
crc[['RNA']] <- CreateAssayObject(counts = gene.activities)
crc <- NormalizeData(
object = crc,
assay = 'RNA',
normalization.method = 'LogNormalize',
scale.factor = median(crc$nCount_RNA)
)
展示特定基因活性得分
在CRC数据集中,识别了以下不同细胞类型的标志性基因:
- EPCAM:上皮细胞的标志
- TREM1:髓系细胞的标志
- PTPRC(也称为CD45):泛免疫细胞的标志
- IL1RL1:嗜碱性粒细胞的标志
- GATA3:T细胞的标志
DefaultAssay(crc) <- 'RNA'
FeaturePlot(
object = crc,
features = c('TREM1', 'EPCAM', "PTPRC", "IL1RL1","GATA3", "KIT"),
pt.size = 0.1,
max.cutoff = 'q95',
ncol = 2
)
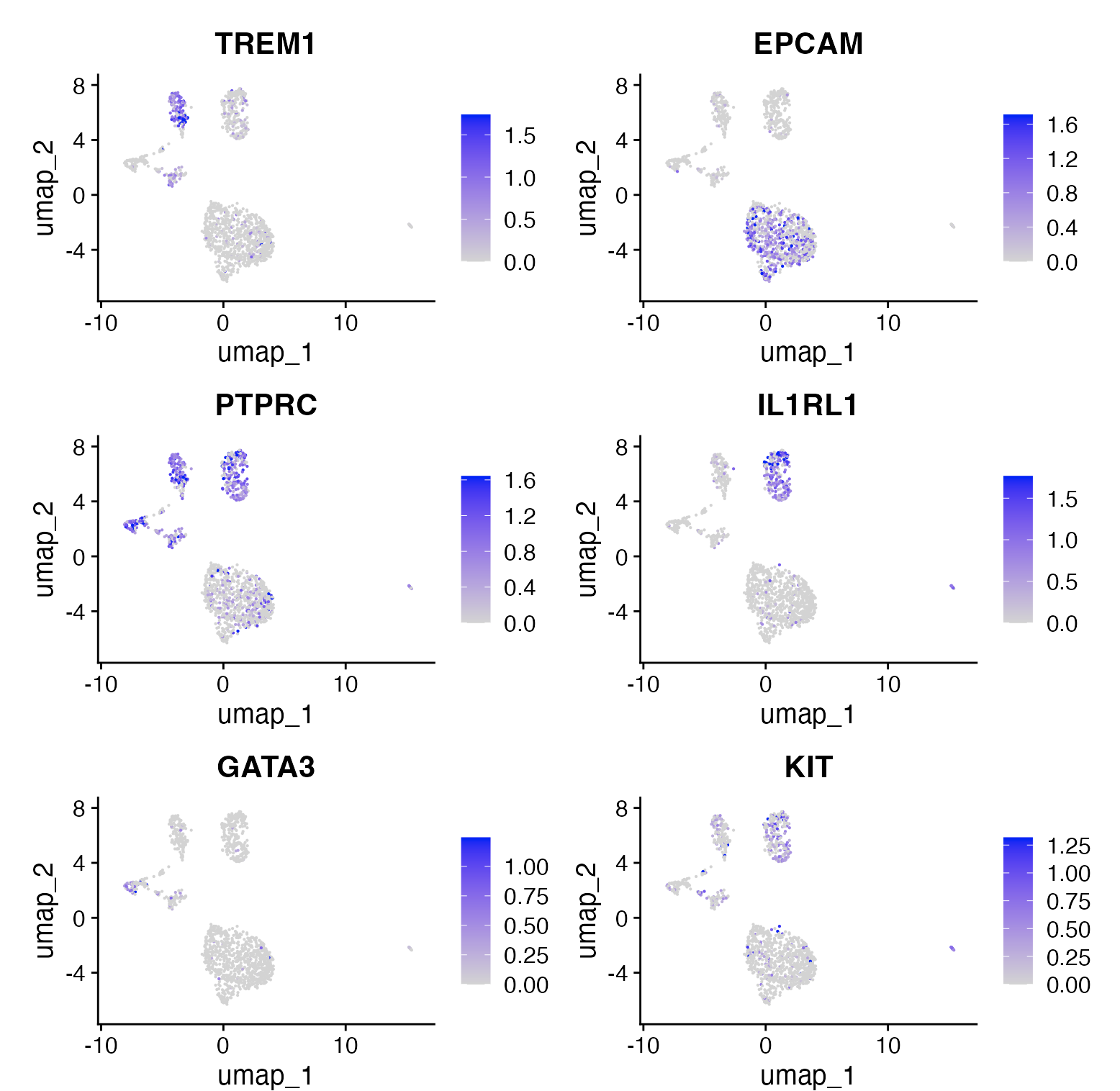
使用这些基因评分值,可以分配簇标识:
crc <- RenameIdents(
object = crc,
'0' = 'Epithelial',
'1' = 'Epithelial',
'2' = 'Basophil',
'3' = 'Myeloid_1',
'4' = 'Myeloid_2',
'5' = 'Tcell'
)
在髓系细胞群中,发现有一个亚群表现出较低的峰值区域片段比例,整体线粒体测序覆盖度也不高,并且其核小体的排列模式与其他亚群有所区别。
p1 <- FeatureScatter(crc, "mtDNA_depth", "pct_reads_in_peaks") + ggtitle("") + scale_x_log10()
p2 <- FeatureScatter(crc, "mtDNA_depth", "nucleosome_signal") + ggtitle("") + scale_x_log10()
p1 + p2 + plot_layout(guides = 'collect')
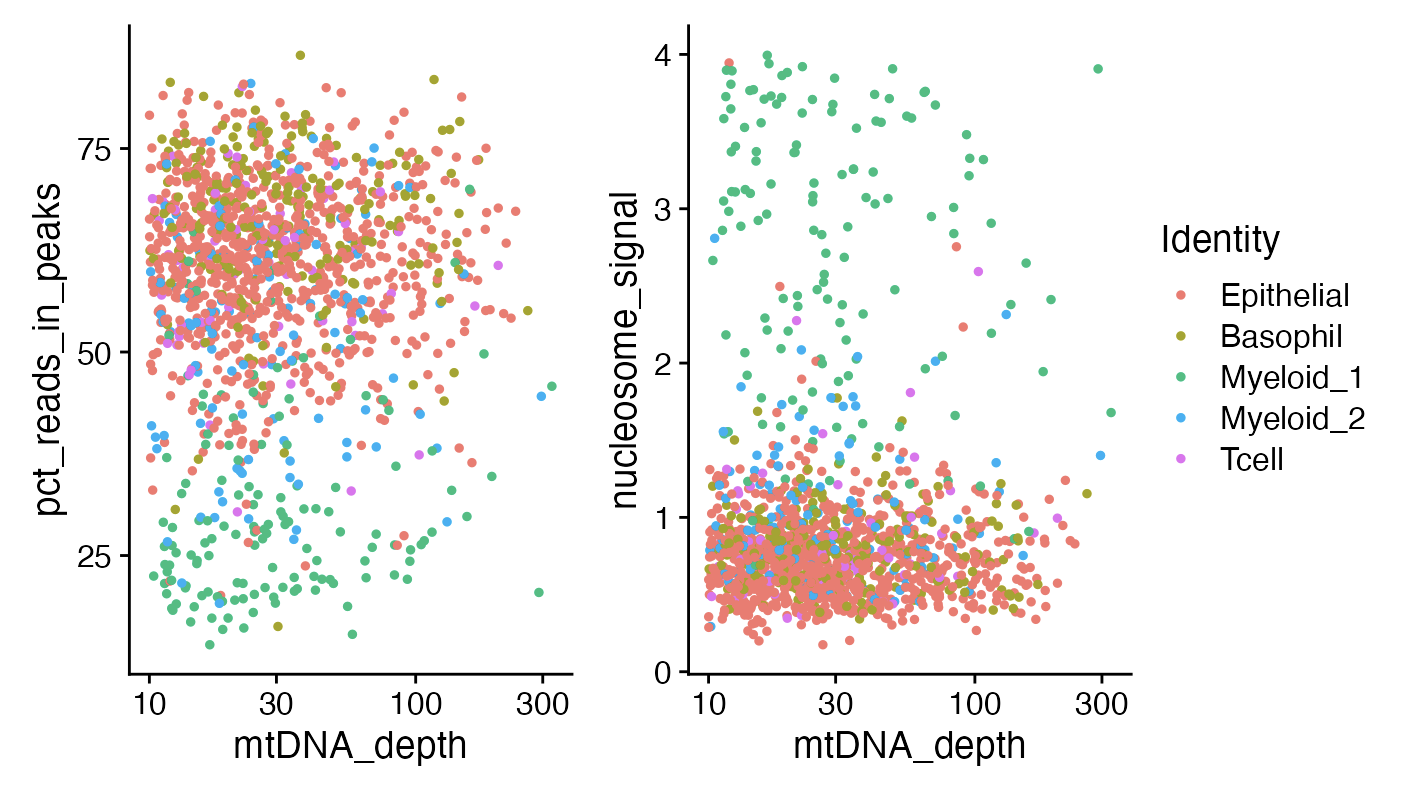
观察到,大多数FRIP值较低的细胞属于髓系1群。这些细胞很可能是肿瘤内的粒细胞,它们在染色质可及性方面的富集程度相对较低。与此相似,这种细胞类型的核染色质包装方式也较为特殊,导致其mtDNA覆盖度略低于髓系2群。
识别有意义的mtDNA变异
接下来,可以确定线粒体基因组中在不同细胞间存在差异的位点,并根据这些变异在细胞中的出现频率将细胞分为不同的克隆型。Signac在识别高质量变异时,采用了原始mtscATAC-seq研究中建立的原则。
variable.sites <- IdentifyVariants(crc, assay = "mito", refallele = mito.data$refallele)
VariantPlot(variants = variable.sites)
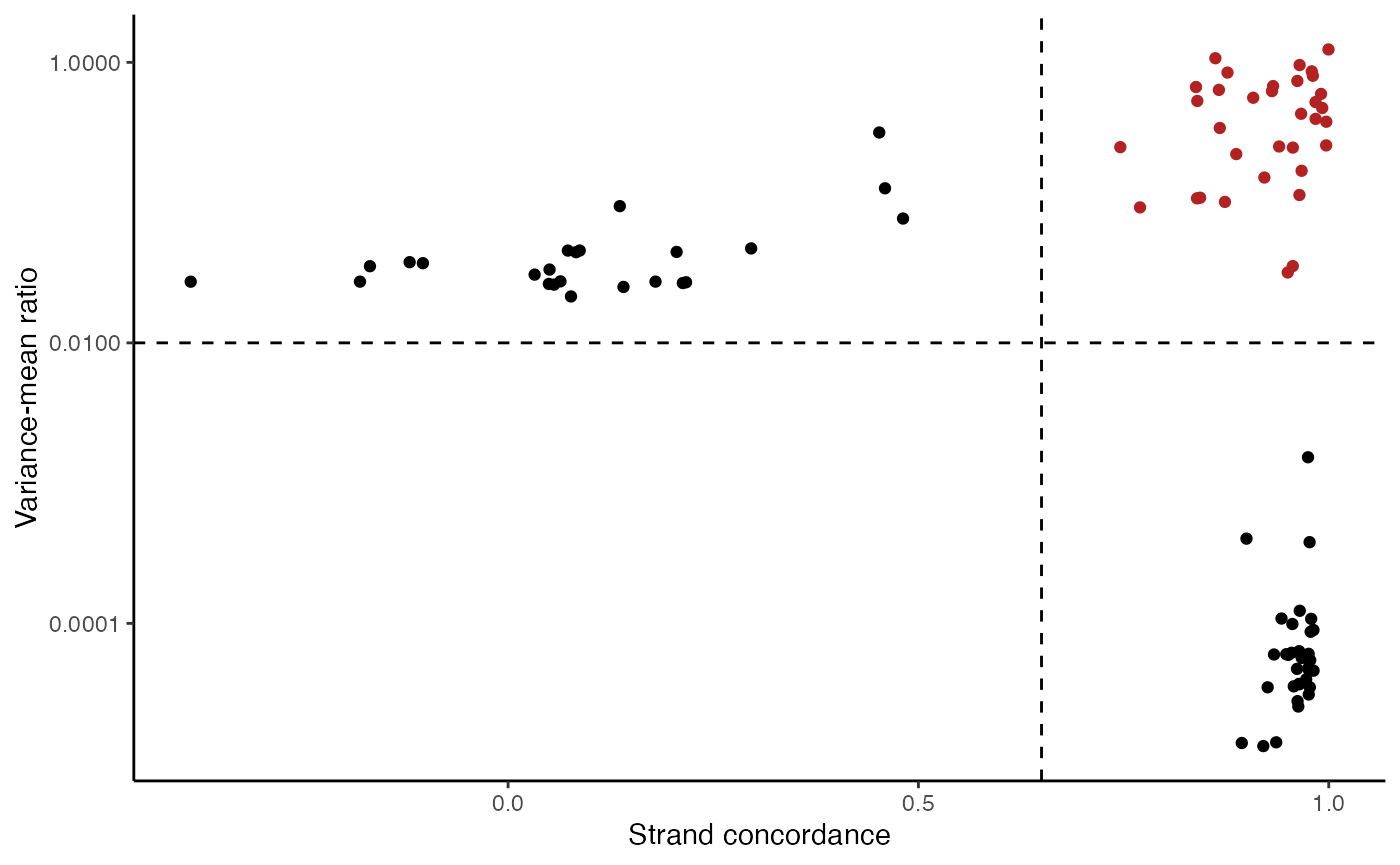
上图清晰地展示了一组变异,这些变异具有较高的变异等位基因比率(VMR)和链一致性。理论上,高链一致性减少了等位基因频率受测序错误影响的可能性(这种错误通常只发生在一个链上,而不是另一个链上,这是由于前导核苷酸的内容和mtDNA基因分型中的一个常见错误)。另一方面,具有高VMR的变异更可能是克隆变异,因为这些变异的替代等位基因倾向于在特定细胞中聚集,而不是均匀分布在所有细胞中,后者则可能表明其他一些技术问题。
注意到,那些具有极低VMR和极高链一致性的变异是这个样本的同源变异。虽然这些变异在某些情况下可能具有研究价值(例如,在供体样本的去复用过程中),但对于推断亚克隆结构,它们并没有太大的用处。
根据这些标准,可以筛选出一组在不同细胞中表现出差异的、具有信息价值的线粒体变异。
# Establish a filtered data frame of variants based on this processing
high.conf <- subset(
variable.sites, subset = n_cells_conf_detected >= 5 &
strand_correlation >= 0.65 &
vmr > 0.01
)
high.conf[,c(1,2,5)]
## position nucleotide mean
## 1227G>A 1227 G>A 0.0090107
## 6081G>A 6081 G>A 0.0031485
## 9804G>A 9804 G>A 0.0035833
## 12889G>A 12889 G>A 0.0217851
## 9728C>T 9728 C>T 0.0150412
## 16147C>T 16147 C>T 0.6582835
## 824T>C 824 T>C 0.0050820
## 2285T>C 2285 T>C 0.0034535
## 16093T>C 16093 T>C 0.0093126
一些关键点值得注意。首先,在12个变异中,有10个在群体中的出现频率不到1%。但是,16147C>T的频率大约为62%。将会认识到这是一个标记上皮细胞的克隆变异。此外,所有检测到的变异都是转换类型(A - G 或 C - T),而不是颠换类型(A - T 或 C - G)。这与对线粒体基因组中这些突变产生方式的认知相吻合。
计算变异等位基因频率
目前拥有存储在mito检测中的每个链的信息,这使得可以评估链的一致性。现在,已经确定了一组高可信度的信息变异,可以使用AlleleFreq()函数,为这些变异创建一个新的检测,包含每个细胞的链折叠等位基因频率计数。
crc <- AlleleFreq(
object = crc,
variants = high.conf$variant,
assay = "mito"
)
crc[["alleles"]]
## Assay data with 9 features for 1359 cells
## First 9 features:
## 1227G>A, 6081G>A, 9804G>A, 12889G>A, 9728C>T, 16147C>T, 824T>C,
## 2285T>C, 16093T>C
展示变异分布
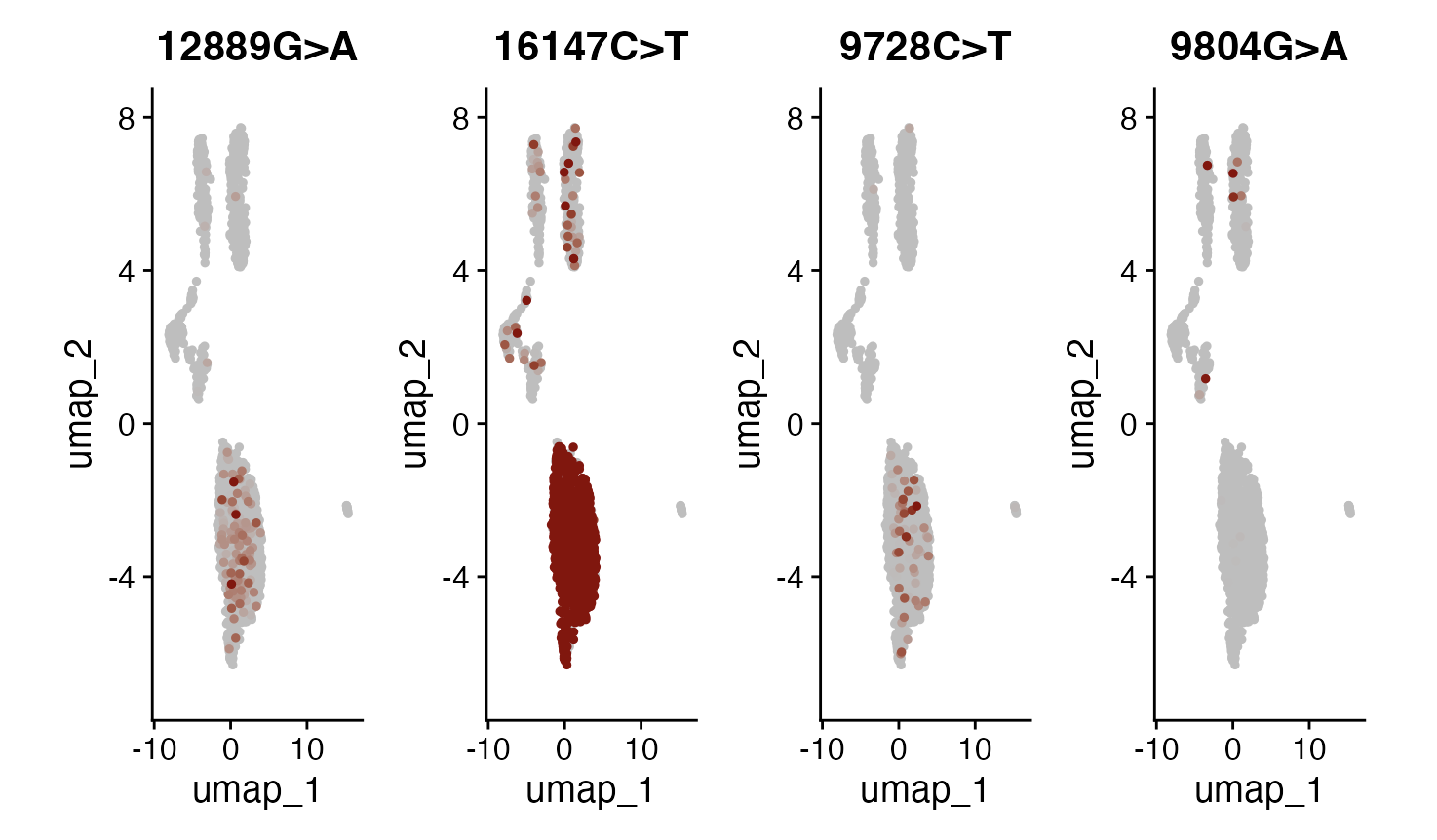
随着等位基因频率被存储为额外的检测数据,可以利用Seurat的标准功能来展示这些频率如何在不同细胞间分布。在这里,通过FeaturePlot()和DoHeatmap()展示了变异的一个子集,以便直观地理解它们的分布情况。
DefaultAssay(crc) <- "alleles"
alleles.view <- c("12889G>A", "16147C>T", "9728C>T", "9804G>A")
FeaturePlot(
object = crc,
features = alleles.view,
order = TRUE,
cols = c("grey", "darkred"),
ncol = 4
) & NoLegend()
DoHeatmap(crc, features = rownames(crc), slot = "data", disp.max = 1) +
scale_fill_viridis_c()
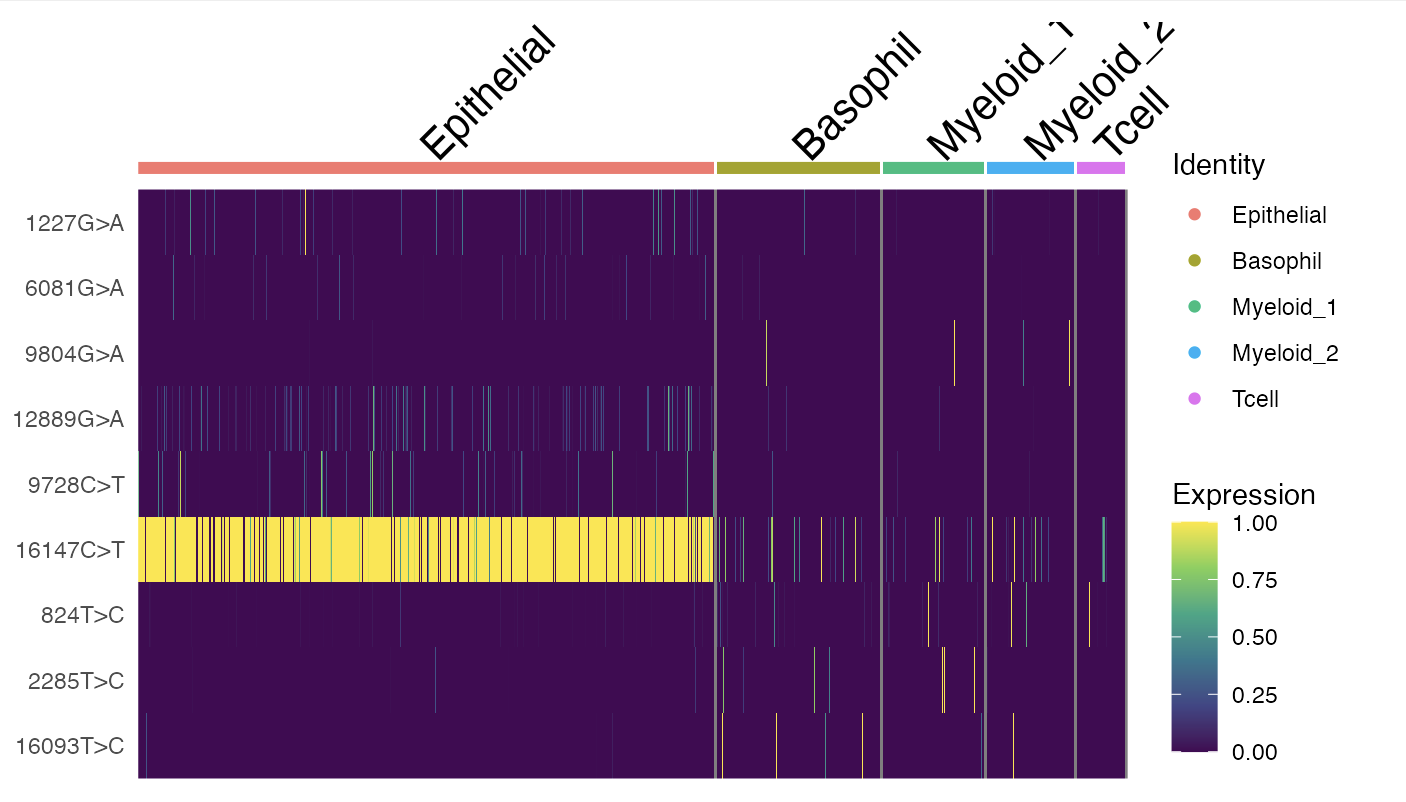
在这些选定的变异中,可以观察到一些有趣的分布模式。变异16147C>T几乎存在于所有的上皮细胞中,而且基本上只出现在上皮细胞内(少数例外情况也对应着UMAP和聚类结果不一致的案例)。这个变异的等位基因频率达到了100%,这强烈表明肿瘤的原始细胞在发生突变后进行了克隆性扩增。接着,至少发现了3个变异——1227G>A、12889G>A和9728C>T,它们主要特定于定义亚克隆的上皮细胞中。另外,包括3244G>A、9804G>A和824T>C在内的其他变异,特别在免疫细胞群体中被发现,这暗示这些变异可能源自一个共同的造血祖细胞(很可能位于骨髓中)。








 苏公网安备32011502012024号
苏公网安备32011502012024号