引言
Visium HD数据是通过在2微米 x 2微米的网格中标记的、具有空间模式的寡核苷酸生成的。由于这种高分辨率下的数据较为稀疏,因此相邻的网格会被合并,形成8微米和16微米的分辨率。虽然10x推荐使用8微米网格的数据进行分析,但Seurat允许同时加载多种不同分辨率的数据,并将它们存储在一个对象中作为多个检测。
在本文中,概述了Seurat支持的一些空间分析工作流程,特别是针对Visium HD数据的分析,包括:
- 无监督聚类
- 识别空间组织区域
- 选择特定的空间区域进行分析
- 与单细胞RNA测序(scRNA-seq)数据的整合
- 对比不同细胞类型在空间上的分布
分析重点是一个来自小鼠大脑的Visium HD数据集。
安装 Seurat 更新
# packages required for Visium HD
install.packages("hdf5r")
install.packages("arrow")
library(Seurat)
library(ggplot2)
library(patchwork)
library(dplyr)
数据加载
- Seurat 能够在同一对象的不同检测中存储多种分辨率的数据。
- 通过设置 bin.size 参数,用户可以选择加载的分辨率,系统默认加载的是8微米和16微米的数据。
- 用户可以通过更改检测来轻松切换不同的分辨率,以适应不同的分析需求。
localdir <- "/brahms/lis/visium_hd/mouse/new_mousebrain/"
object <- Load10X_Spatial(data.dir = localdir, bin.size = c(8, 16))
# Setting default assay changes between 8um and 16um binning
Assays(object)
DefaultAssay(object) <- "Spatial.008um"
vln.plot <- VlnPlot(object, features = "nCount_Spatial.008um", pt.size = 0) + theme(axis.text = element_text(size = 4)) + NoLegend()
count.plot <- SpatialFeaturePlot(object, features = "nCount_Spatial.008um") + theme(legend.position = "right")
# note that many spots have very few counts, in-part
# due to low cellular density in certain tissue regions
vln.plot | count.plot

归一化
采用了对空间数据进行常规的对数转换标准化方法。我们指出,针对空间数据的最佳标准化技术仍在不断研究和评估之中。我们推荐用户深入阅读Phipson/Davis实验室以及Fan实验室发表的论文,以获取有关空间数据标准化潜在陷阱的更多信息。
# normalize both 8um and 16um bins
DefaultAssay(object) <- "Spatial.008um"
object <- NormalizeData(object)
DefaultAssay(object) <- "Spatial.016um"
object <- NormalizeData(object)
基因表达可视化
- 调整 pt.size.factor(默认设置为 1.2)有助于可视化此 HD 数据集中的分子和组织学信息
- 您还可以调整形状和描边(轮廓)参数以实现可视化
# switch spatial resolution to 16um from 8um
DefaultAssay(object) <- "Spatial.016um"
p1 <- SpatialFeaturePlot(object, features = "Rorb") + ggtitle("Rorb expression (16um)")
# switch back to 8um
DefaultAssay(object) <- "Spatial.008um"
p2 <- SpatialFeaturePlot(object, features = "Hpca") + ggtitle("Hpca expression (8um)")
p1 | p2

无监督聚类
尽管常规的单细胞RNA测序聚类流程同样适用于空间数据集,我们发现在使用Visium HD数据集时,Seurat v5的草图聚类方法在性能上有所提升,特别是在识别那些数量稀少且在空间上有特定分布的细胞群组时。
在本例中,我们对Visium HD数据集进行了草图化处理,对这些抽样得到的细胞进行了聚类分析,并将得到的聚类结果映射回整个数据集。
# note that data is already normalized
DefaultAssay(object) <- "Spatial.008um"
object <- FindVariableFeatures(object)
object <- ScaleData(object)
# we select 50,0000 cells and create a new 'sketch' assay
object <- SketchData(
object = object,
ncells = 50000,
method = "LeverageScore",
sketched.assay = "sketch"
)
# switch analysis to sketched cells
DefaultAssay(object) <- "sketch"
# perform clustering workflow
object <- FindVariableFeatures(object)
object <- ScaleData(object)
object <- RunPCA(object, assay = "sketch", reduction.name = "pca.sketch")
object <- FindNeighbors(object, assay = "sketch", reduction = "pca.sketch", dims = 1:50)
object <- FindClusters(object, cluster.name = "seurat_cluster.sketched", resolution = 3)
object <- RunUMAP(object, reduction = "pca.sketch", reduction.name = "umap.sketch", return.model = T, dims = 1:50)
接下来,我们可以利用ProjectData函数,将从5万个草图化细胞中得到的聚类标签和降维分析(主成分分析PCA和统一流形近似UMAP)结果,映射到整个数据集中。
映射完成后,在生成的对象里,所有细胞的相关信息将如下存储:
- 聚类标签将被记录在对象的
object$seurat_cluster.projected属性中。
- 映射后的PCA降维结果将存放于对象的
object[["pca.008um"]]字段。
- 映射后的UMAP降维结果将存放于对象的
object[["umap.sketch"]]字段。
object <- ProjectData(
object = object,
assay = "Spatial.008um",
full.reduction = "full.pca.sketch",
sketched.assay = "sketch",
sketched.reduction = "pca.sketch",
umap.model = "umap.sketch",
dims = 1:50,
refdata = list(seurat_cluster.projected = "seurat_cluster.sketched")
)
我们可以对草图化处理后的细胞进行聚类结果的可视化展示,同时也可以对整个数据集的聚类结果进行映射后的可视化分析。
DefaultAssay(object) <- "sketch"
Idents(object) <- "seurat_cluster.sketched"
p1 <- DimPlot(object, reduction = "umap.sketch", label = F) + ggtitle("Sketched clustering (50,000 cells)") + theme(legend.position = "bottom")
# switch to full dataset
DefaultAssay(object) <- "Spatial.008um"
Idents(object) <- "seurat_cluster.projected"
p2 <- DimPlot(object, reduction = "full.umap.sketch", label = F) + ggtitle("Projected clustering (full dataset)") + theme(legend.position = "bottom")
p1 | p2

当然,我们现在还可以根据空间位置来可视化无监督集群。请注意,运行 SpatialDimPlot(object, Interactive = TRUE) 还可以实现交互式可视化和探索。
SpatialDimPlot(object, label = T, repel = T, label.size = 4)

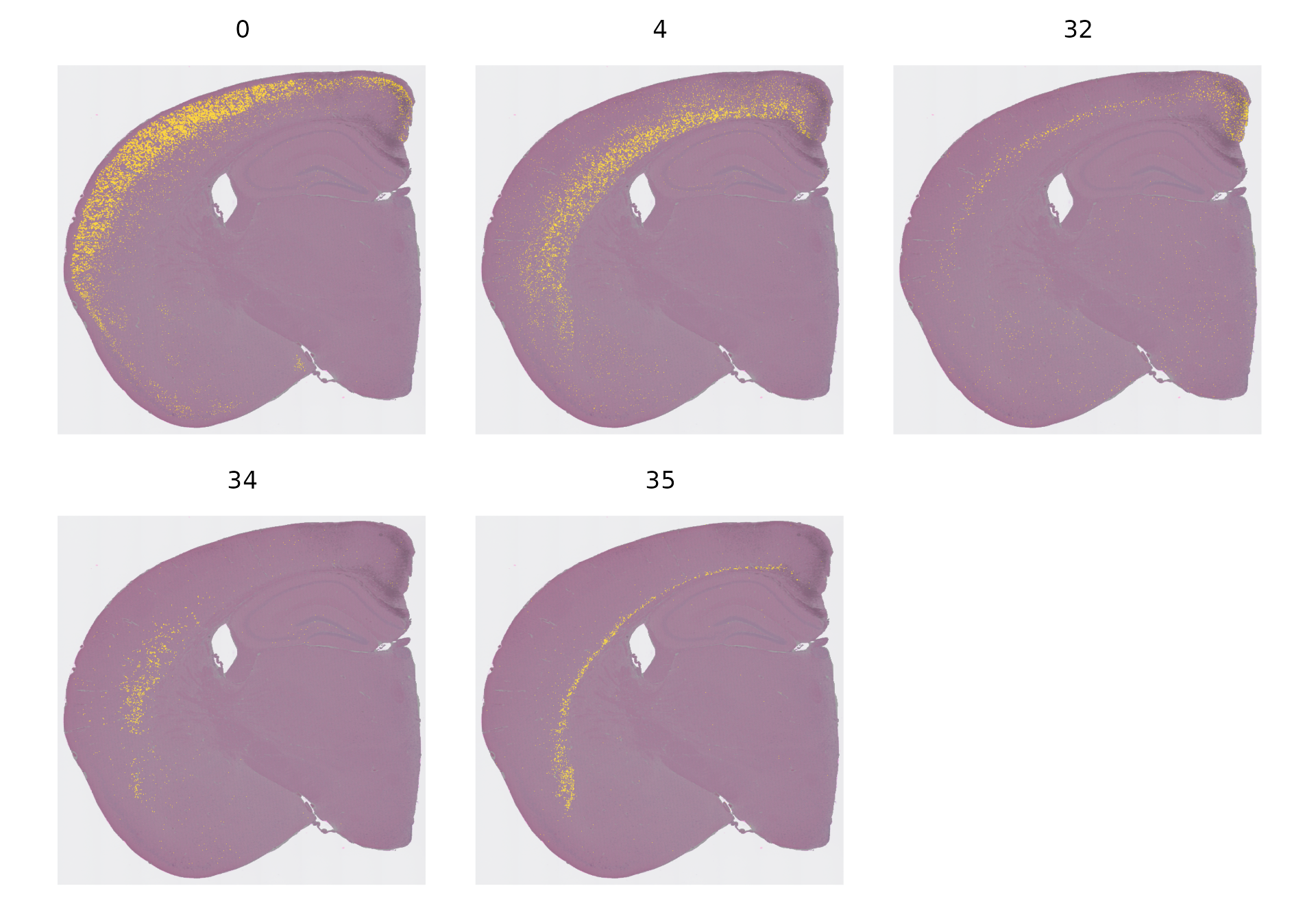
当面对众多不同的细胞聚类——有些聚类的空间分布受到限制,而有些则分布较为混杂——时,尝试在一张图上展示它们的空间分布可能会让人难以理解。为了解决这个问题,我们通常会单独展示每个聚类的空间分布。例如,我们在下面的图中特别标注了几个聚类的空间位置,这些聚类恰好代表了大脑皮层的不同层次。
Idents(object) <- "seurat_cluster.projected"
cells <- CellsByIdentities(object, idents = c(0, 4, 32, 34, 35))
p <- SpatialDimPlot(object,
cells.highlight = cells[setdiff(names(cells), "NA")],
cols.highlight = c("#FFFF00", "grey50"), facet.highlight = T, combine = T
) + NoLegend()
p
我们还可以找到并可视化每个簇的顶级基因表达标记:
# Crete downsampled object to make visualization either
DefaultAssay(object) <- "Spatial.008um"
Idents(object) <- "seurat_cluster.projected"
object_subset <- subset(object, cells = Cells(object[["Spatial.008um"]]), downsample = 1000)
# Order clusters by similarity
DefaultAssay(object_subset) <- "Spatial.008um"
Idents(object_subset) <- "seurat_cluster.projected"
object_subset <- BuildClusterTree(object_subset, assay = "Spatial.008um", reduction = "full.pca.sketch", reorder = T)
markers <- FindAllMarkers(object_subset, assay = "Spatial.008um", only.pos = TRUE)
markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
object_subset <- ScaleData(object_subset, assay = "Spatial.008um", features = top5$gene)
p <- DoHeatmap(object_subset, assay = "Spatial.008um", features = top5$gene, size = 2.5) + theme(axis.text = element_text(size = 5.5)) + NoLegend()
p

鉴定具有空间特征的组织区域
与之前独立分析每个样本格子的方法不同,空间数据允许我们不仅根据细胞的邻近环境,还可以根据它们更广阔的空间背景来定义细胞。
if (!requireNamespace("Banksy", quietly = TRUE)) {
remotes::install_github("prabhakarlab/Banksy@devel")
}
library(SeuratWrappers)
library(Banksy)
在启动 BANKSY 工具之前,用户需要考虑两个关键的模型参数:
- k_geom:指局部邻域的规模大小。设置较大的值会导致识别出更大的组织区域。
- lambda:表示邻域对结果的影响程度。较高的值有助于生成在空间上更加连贯一致的区域划分。
RunBanksy 函数会生成一个新的 BANKSY 分析结果集,这个结果集可以用于后续的降维处理和细胞聚类分析。
object <- RunBanksy(object,
lambda = 0.8, verbose = TRUE,
assay = "Spatial.008um", slot = "data", features = "variable",
k_geom = 50
)
DefaultAssay(object) <- "BANKSY"
object <- RunPCA(object, assay = "BANKSY", reduction.name = "pca.banksy", features = rownames(object), npcs = 30)
object <- FindNeighbors(object, reduction = "pca.banksy", dims = 1:30)
object <- FindClusters(object, cluster.name = "banksy_cluster", resolution = 0.5)
Idents(object) <- "banksy_cluster"
p <- SpatialDimPlot(object, group.by = "banksy_cluster", label = T, repel = T, label.size = 4)
p

与无监督聚类一样,我们可以单独突出显示每个组织域的空间位置:
banksy_cells <- CellsByIdentities(object)
p <- SpatialDimPlot(object, cells.highlight = banksy_cells[setdiff(names(banksy_cells), "NA")], cols.highlight = c("#FFFF00", "grey50"), facet.highlight = T, combine = T) + NoLegend()
p









 苏公网安备32011502012024号
苏公网安备32011502012024号