引言
在这篇指南中,我们介绍了Seurat的一个新扩展功能,用以分析新型的空间解析数据,将重点介绍由不同成像技术生成的三个公开数据集。
- Vizgen MERSCOPE(用于小鼠大脑研究)
- Nanostring CosMx空间分子成像仪(用于FFPE人类肺组织)
- Akoya CODEX(用于人类淋巴结研究)
首先,我们将加载完成本指南所需的相关软件包。
library(Seurat)
library(future)
plan("multisession", workers = 10)
library(ggplot2)
小鼠大脑:Vizgen MERSCOPE
这个数据集是通过Vizgen MERSCOPE系统生成的,该系统采用了MERFISH技术。整个数据集包含了九个样本(小鼠大脑的三个完整横截面,每个横截面有三个生物学重复样本)。基因面板涵盖了483个基因目标,包括了已知的典型细胞类型标志物、非感觉型G蛋白偶联受体(GPCRs)和受体酪氨酸激酶(RTKs)。
在本指南中,我们分析了其中一个样本——第二切片的第一个生物学重复样本。在每个细胞中检测到的转录本数量平均为206。
首先,我们导入数据集并构建了一个Seurat对象。
我们使用了我们自己编写的LoadVizgen()函数来读取Vizgen分析流程的结果。生成的Seurat对象包含了以下信息:一个计数矩阵,记录了每个细胞中483个转录本的观察分子数。这个矩阵在功能上与单细胞RNA测序中的计数矩阵相似,并且默认情况下存储在Seurat对象的RNA分析模块中。
vizgen.obj <- LoadVizgen(data.dir = "/brahms/hartmana/vignette_data/vizgen/s2r1/", fov = "s2r1")
接下来的信息特定于成像分析,并存储在生成的 Seurat 对象的图像槽中:
head(GetTissueCoordinates(vizgen.obj[["s2r1"]][["centroids"]]))
## x y cell
## 1 638.5640 4594.216 149164679103246548309819743981609972453
## 2 593.8034 4516.240 215843146921706462965382248182021894607
## 3 597.3134 4566.676 230248905804673613678286091156141465134
## 4 613.2434 4609.498 237155298815097057940587033798543926454
## 5 609.1934 4603.180 256099454901769634241742157204636917386
## 6 623.8814 4642.708 52442222147121971758529793775250916001
- 细胞边界划分:用于界定每个独立细胞轮廓的一系列空间坐标点,形成多边形以表示细胞的边界。
head(GetTissueCoordinates(vizgen.obj[["s2r1"]][["segmentation"]]))
## x y cell
## 1 644.0774 4589.022 149164679103246548309819743981609972453
## 2 643.9694 4589.022 149164679103246548309819743981609972453
## 3 643.8614 4589.022 149164679103246548309819743981609972453
## 4 643.7642 4588.924 149164679103246548309819743981609972453
## 5 643.7534 4588.914 149164679103246548309819743981609972453
## 6 643.6454 4588.914 149164679103246548309819743981609972453
- 分子定位信息:记录了在多重单分子荧光原位杂交(smFISH)实验过程中发现的每一个分子的空间位置。
head(FetchData(vizgen.obj[["s2r1"]][["molecules"]], vars = "Chrm1"))
## x y molecule
## 1 577.3373 4205.977 Chrm1
## 2 600.0218 3917.781 Chrm1
## 3 508.2736 3934.063 Chrm1
## 4 630.7590 3948.586 Chrm1
## 5 635.1143 3969.567 Chrm1
## 6 582.7043 4021.577 Chrm1
预处理和无监督分析
我们首先进行常规的无监督聚类分析,将数据集初步处理为单细胞RNA测序(scRNA-seq)实验。在标准化过程中,我们采用了基于SCTransform的方法,并对默认的裁剪参数进行了微调,以减少smFISH实验中偶尔出现的异常值对我们分析结果的干扰。完成标准化后,我们便可以进行数据的降维处理和聚类分析。
vizgen.obj <- SCTransform(vizgen.obj, assay = "Vizgen", clip.range = c(-10, 10))
vizgen.obj <- RunPCA(vizgen.obj, npcs = 30, features = rownames(vizgen.obj))
vizgen.obj <- RunUMAP(vizgen.obj, dims = 1:30)
vizgen.obj <- FindNeighbors(vizgen.obj, reduction = "pca", dims = 1:30)
vizgen.obj <- FindClusters(vizgen.obj, resolution = 0.3)
然后,我们可以在 UMAP 空间(使用 DimPlot())中可视化聚类结果,或者使用 ImageDimPlot() 覆盖在图像上。
DimPlot(vizgen.obj, reduction = "umap")
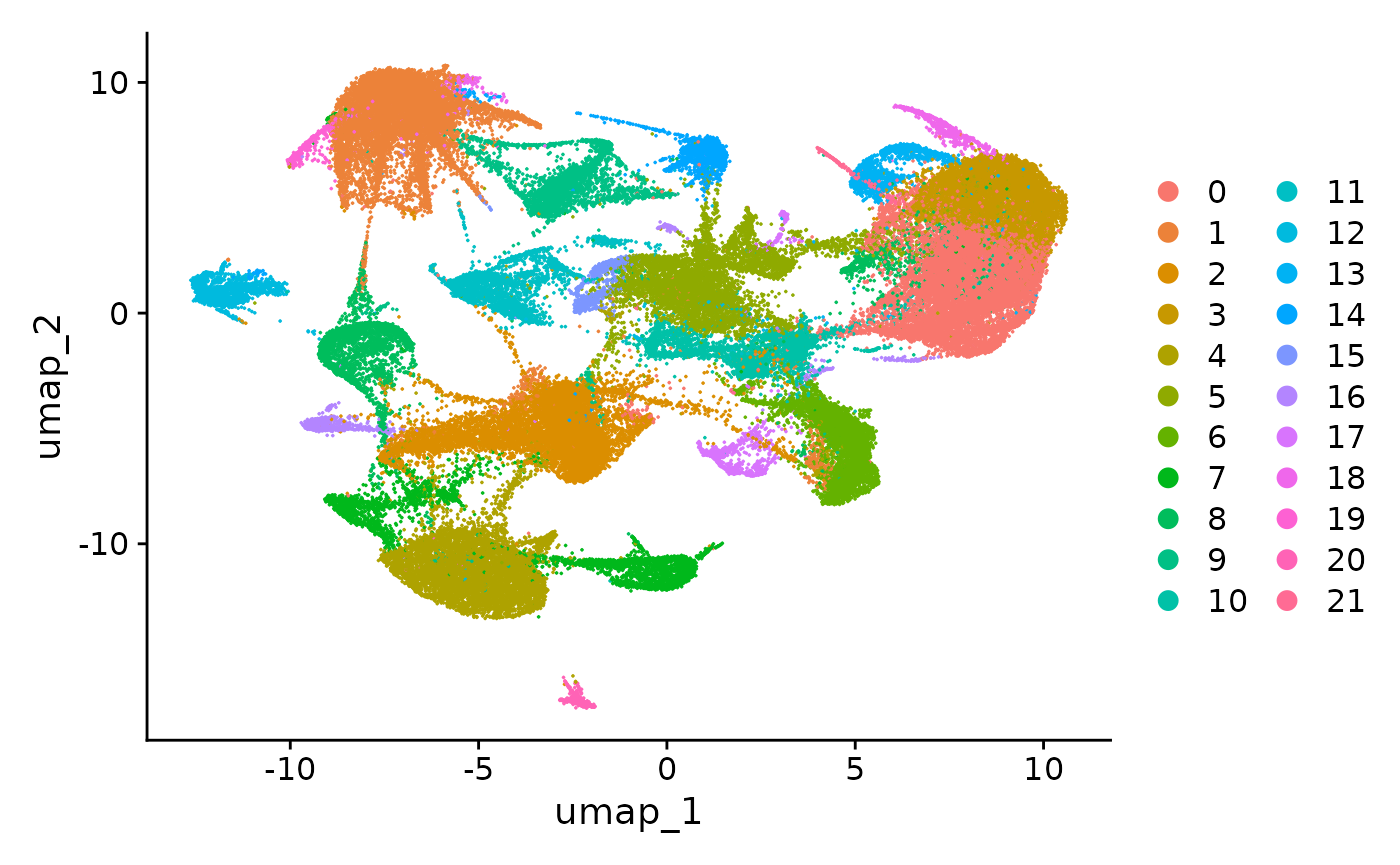
ImageDimPlot(vizgen.obj, fov = "s2r1", cols = "polychrome", axes = TRUE)

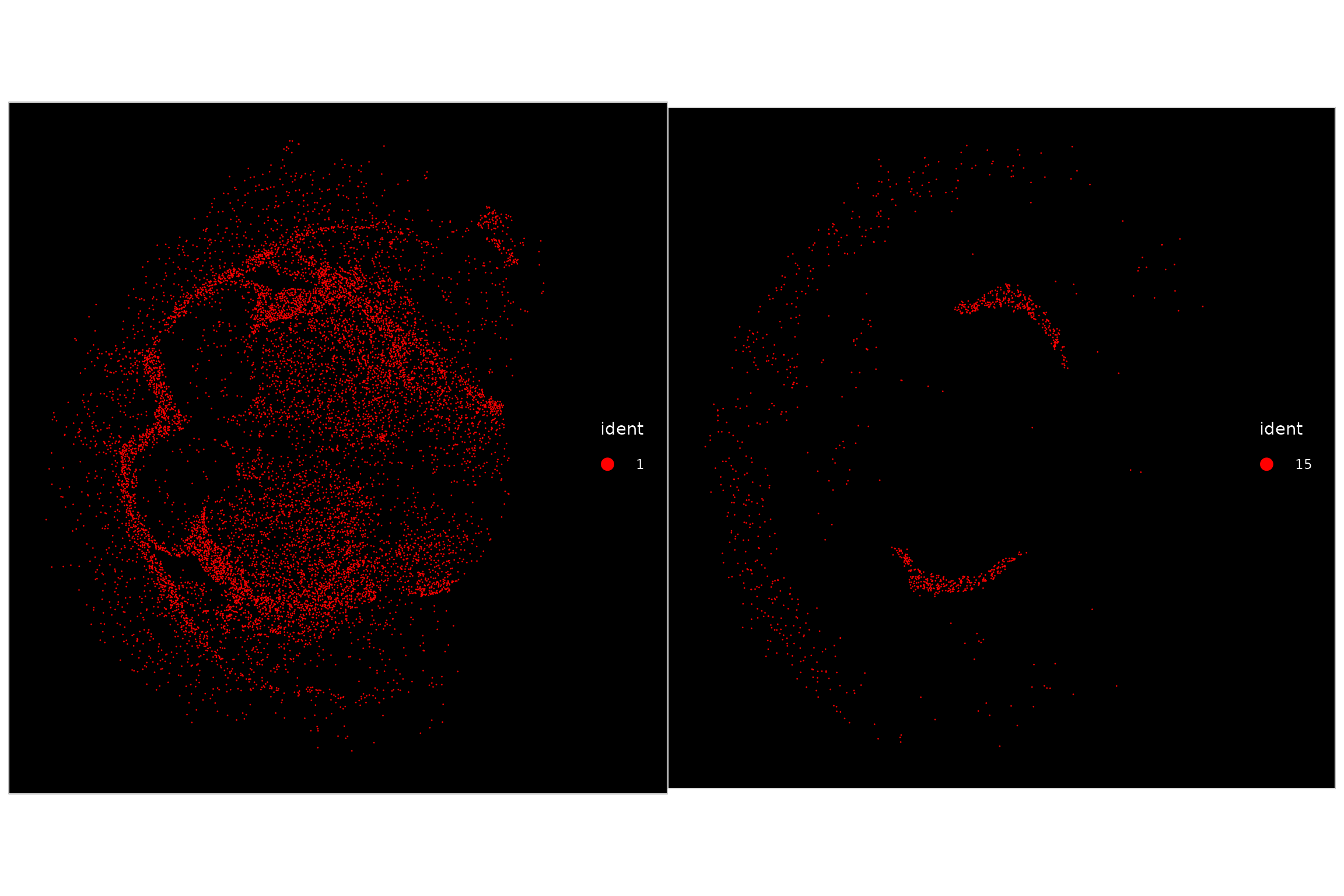
由于在一起查看单个簇时很难可视化它们的空间定位模式,因此我们可以突出显示属于特定簇的所有细胞:
p1 <- ImageDimPlot(vizgen.obj, fov = "s2r1", cols = "red", cells = WhichCells(vizgen.obj, idents = 1))
p2 <- ImageDimPlot(vizgen.obj, fov = "s2r1", cols = "red", cells = WhichCells(vizgen.obj, idents = 15))
p1 + p2
我们能够识别每个独立聚类的特征标志物,并展示它们在空间上的表达分布。通过使用ImageFeaturePlot()函数,我们可以根据单个基因的表达量来对细胞进行着色,这与FeaturePlot()函数的作用相似,都是为了在二维平面上展示基因表达的分布情况。考虑到MERFISH技术能够对单个分子进行成像,我们还能够在图像上直接观察到每个分子的具体位置。
p1 <- ImageFeaturePlot(vizgen.obj, features = "Slc17a7")
p2 <- ImageDimPlot(vizgen.obj, molecules = "Slc17a7", nmols = 10000, alpha = 0.3, mols.cols = "red")
p1 + p2
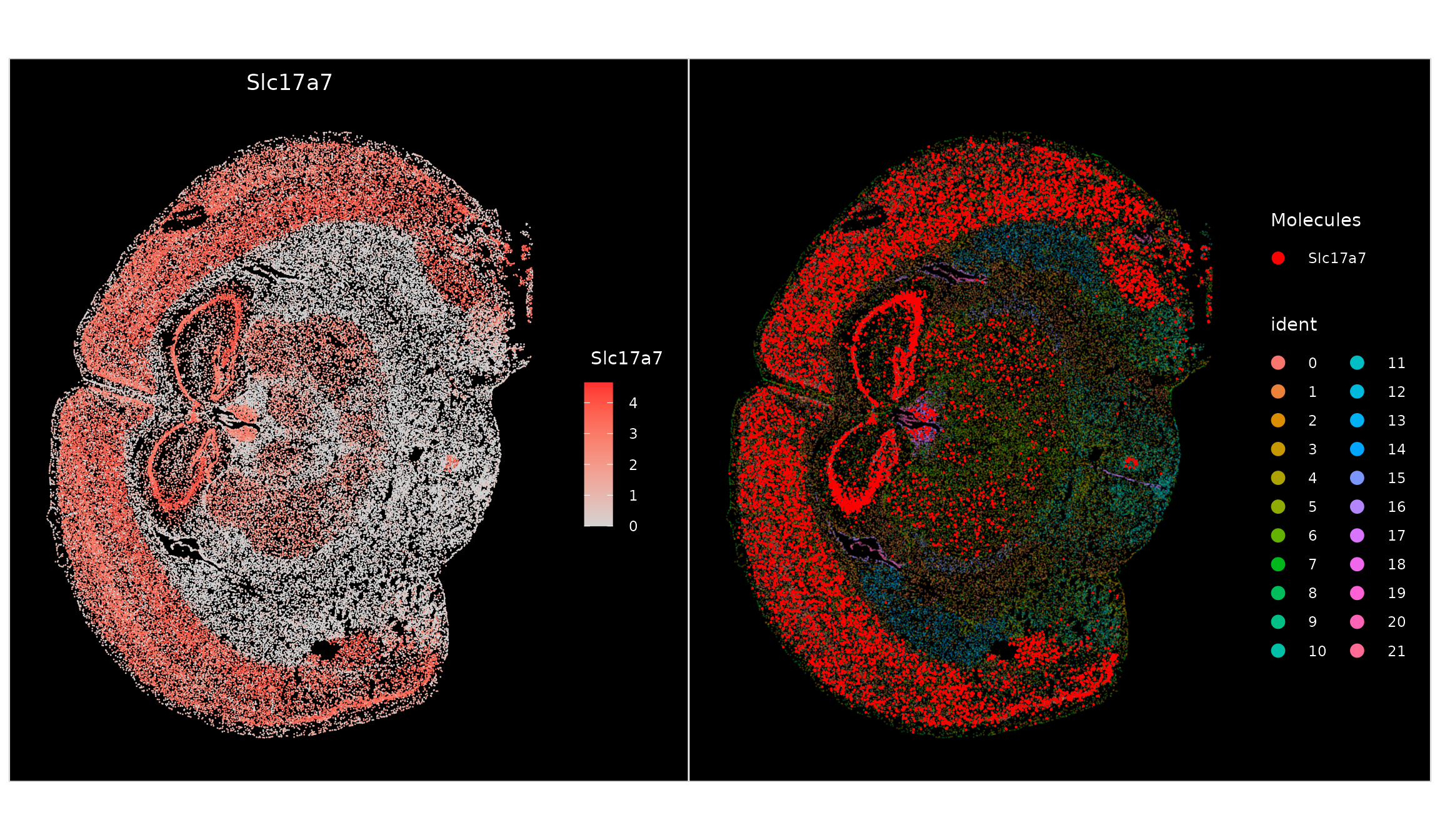
请注意,我们可以通过设置 nmols参数来降低图中展示的分子数量,这样可以避免因分子过多而导致的重叠问题。此外,您还可以调整 mols.size(分子大小)、mols.cols(分子颜色)和 mols.alpha(分子透明度)等参数,以进一步优化图表的展示效果。
在图表上绘制分子对于展示同一图表中多个基因的共表达情况尤为有用。
p1 <- ImageDimPlot(vizgen.obj, fov = "s2r1", alpha = 0.3, molecules = c("Slc17a7", "Olig1"), nmols = 10000)
markers.14 <- FindMarkers(vizgen.obj, ident.1 = "14")
p2 <- ImageDimPlot(vizgen.obj, fov = "s2r1", alpha = 0.3, molecules = rownames(markers.14)[1:4],
nmols = 10000)
p1 + p2

最新版的Seurat空间分析框架提供了两种显示细胞的方式:
- 一种是将细胞作为单独的点来处理,另一种是展示细胞的边界(即细胞的轮廓)。默认情况下,Seurat不显示细胞的轮廓,而是将每个细胞简化为一个点(称为“中心点”),这样做可以提高在大面积区域绘图时的效率,因为在这些区域中,细胞的边界细节往往难以辨认。
- 我们可以通过放大来聚焦组织的特定区域,从而形成一个新的观察视野。比如,我们可以放大到包含海马体的区域进行观察。放大后,我们可以通过设置DefaultBoundary()来展示细胞的边界。此外,为了提高绘图的效率,我们还可以选择“简化”这些细胞边界,即减少每个多边形的边数。
# create a Crop
cropped.coords <- Crop(vizgen.obj[["s2r1"]], x = c(1750, 3000), y = c(3750, 5250), coords = "plot")
# set a new field of view (fov)
vizgen.obj[["hippo"]] <- cropped.coords
# visualize FOV using default settings (no cell boundaries)
p1 <- ImageDimPlot(vizgen.obj, fov = "hippo", axes = TRUE, size = 0.7, border.color = "white", cols = "polychrome",
coord.fixed = FALSE)
# visualize FOV with full cell segmentations
DefaultBoundary(vizgen.obj[["hippo"]]) <- "segmentation"
p2 <- ImageDimPlot(vizgen.obj, fov = "hippo", axes = TRUE, border.color = "white", border.size = 0.1,
cols = "polychrome", coord.fixed = FALSE)
# simplify cell segmentations
vizgen.obj[["hippo"]][["simplified.segmentations"]] <- Simplify(coords = vizgen.obj[["hippo"]][["segmentation"]],
tol = 3)
DefaultBoundary(vizgen.obj[["hippo"]]) <- "simplified.segmentations"
# visualize FOV with simplified cell segmentations
DefaultBoundary(vizgen.obj[["hippo"]]) <- "simplified.segmentations"
p3 <- ImageDimPlot(vizgen.obj, fov = "hippo", axes = TRUE, border.color = "white", border.size = 0.1,
cols = "polychrome", coord.fixed = FALSE)
p1 + p2 + p3

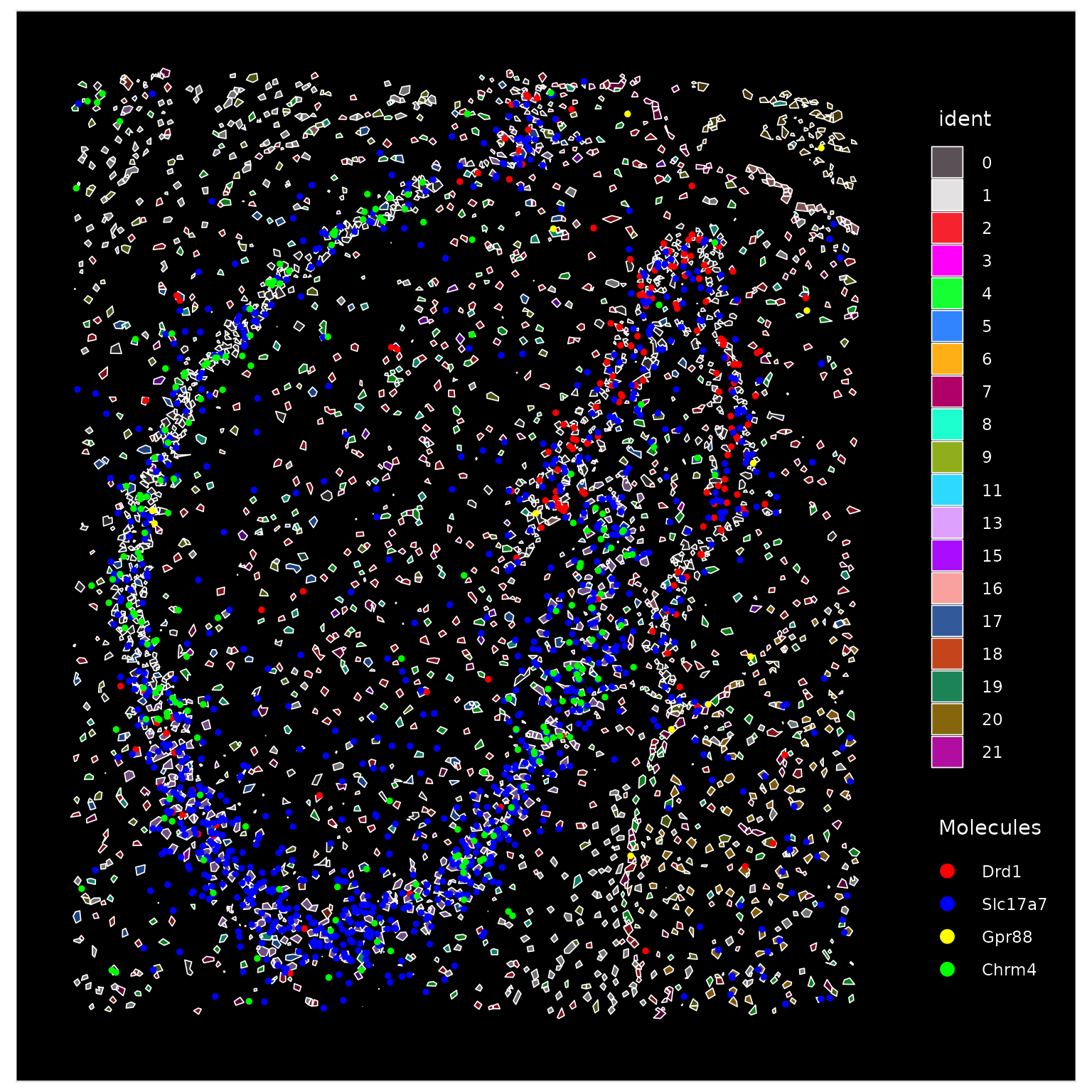
我们可以在放大后以更高分辨率可视化绘制的单个分子
# Since there is nothing behind the segmentations, alpha will slightly mute colors
ImageDimPlot(vizgen.obj, fov = "hippo", molecules = rownames(markers.14)[1:4], cols = "polychrome",
mols.size = 1, alpha = 0.5, mols.cols = c("red", "blue", "yellow", "green"))








 苏公网安备32011502012024号
苏公网安备32011502012024号