泛基因家族分析通常会分析 kaks 和 核苷酸多样性,之前的推文介绍过使用wgd这个软件计算dnds,这个软件调用的是codeml。计算dnds的时候可以选择把比对好的文件保存下来,直接用这个保存好的比对文件计算核苷酸多样性。codeml计算kaks的代码
wgd ksd --n_threads 8 02.mcl 02.cds/ZH13.v2.CDS.fasta --preserve
02.mcl 是某个基因家族所有序列id
每行是一个基因家族,序列id用制表符分割
ZH13.v2.CDS.fasta 是所有的cds序列
保存的比对文件
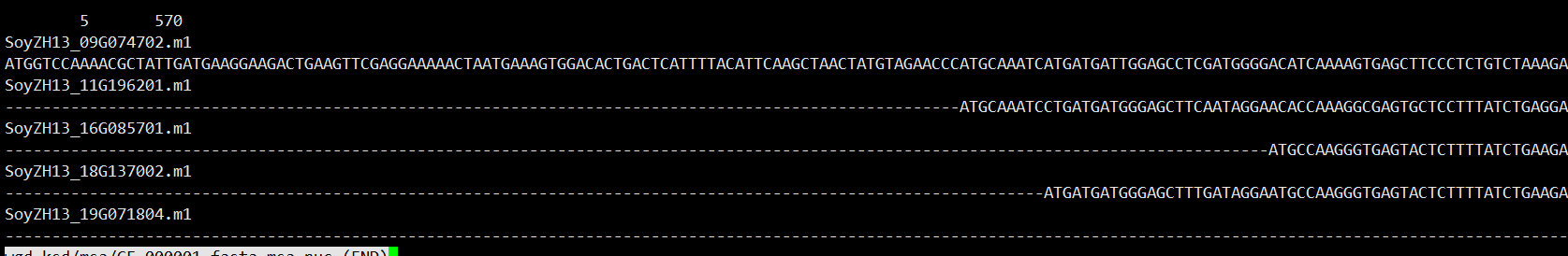
这个比对格式忘记叫什么格式了
R语言里 pegas 包里有一个函数 nuc.div()函数可以直接计算核苷酸多样性
这个函数的输入是 ape包 read.dna()这个函数读取fasta格式的比对序列,读取以后是个DNAbin格式
如何用R语言的函数把wgd 保存的比对文件读入并保存成 DNAbin这个格式 没有找到现成的函数
ape 这个R包里有函数 as.DNAbin() 可以把矩阵转换成 DNAbin的格式
比如
library(tidyverse)
c("A","T","C","G","A","A","C","G") %>%
matrix(nrow = 2,byrow = TRUE) %>%
ape::as.DNAbin()
library(tidyverse)
c("A","T","C","G","A","A","C","G") %>%
matrix(nrow = 2,byrow = TRUE) %>%
ape::as.DNAbin() %>%
pegas::nuc.div()
把上面提到的格式数据读取进来转换成一个矩阵就行了
library(tidyverse)
read_lines("D:/Jupyter/panGeneFamily/GF_000001.fasta.msa.nuc",
skip = 1) %>%
matrix(ncol=2,byrow=TRUE) %>%
as.data.frame() %>%
pull(V2) %>%
str_split(pattern = "") %>%
unlist() %>%
matrix(nrow = 5,byrow = TRUE) %>%
as.DNAbin()
欢迎大家关注我的公众号
小明的数据分析笔记本
小明的数据分析笔记本 公众号 主要分享:1、R语言和python做数据分析和数据可视化的简单小例子;2、园艺植物相关转录组学、基因组学、群体遗传学文献阅读笔记;3、生物信息学入门学习资料及自己的学习笔记!





 苏公网安备32011502012024号
苏公网安备32011502012024号