简介
单细胞测序数据集的整合(例如跨实验批次、供体或条件)通常是 scRNA-seq 工作流程中的重要步骤。整合分析可以帮助匹配跨数据集的共享细胞类型和状态,这可以提高统计能力,最重要的是,促进跨数据集的准确比较分析。
在 Seurat 的早期版本中,引入了整合分析方法,包括“anchor-based”的整合工作流程。许多实验室还发布了用于整合分析的强大且开创性的方法,包括 Harmony 和 scVI 。
目的
以下教程旨在使用 Seurat 对复杂细胞类型进行的各种比较分析。在这里,我们解决了几个关键目标:
- 识别两个数据集中都存在的细胞亚群
- 获得在对照细胞和受刺激细胞中均保守的细胞类型标记
- 比较数据集以发现细胞类型对刺激的特异性反应
对象创建
为了方便起见,我们通过 SeuratData 包分发此数据集。
library(Seurat)
library(SeuratData)
library(patchwork)
# install dataset
InstallData("ifnb")
该对象包含来自两种条件的人类 PBMC 的数据:干扰素刺激细胞和对照细胞。我们的目标是将这两种条件整合在一起,以便我们可以共同识别数据集中的细胞亚群,然后探索每个组在不同条件下的差异。
在 Seurat 的早期版本中,我们要求将数据表示为两个不同的 Seurat 对象。在 Seurat v5 中,我们将所有数据保存在一个对象中,但只是将其分成多个“layers”。
# load dataset
ifnb <- LoadData("ifnb")
# split the RNA measurements into two layers one for control cells, one for stimulated cells
ifnb[["RNA"]] <- split(ifnb[["RNA"]], f = ifnb$stim)
ifnb
不整合分析
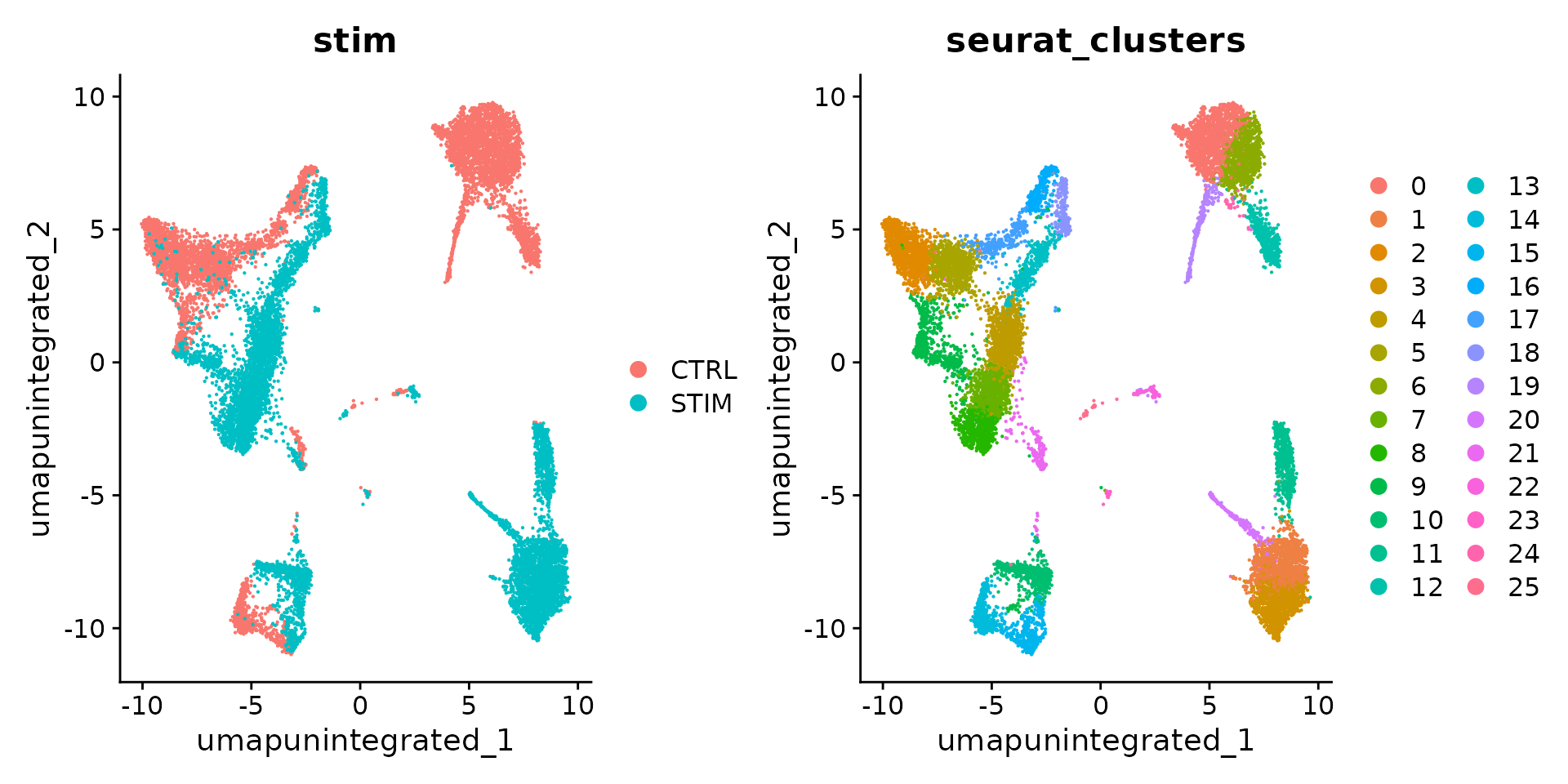
我们可以先分析数据集而不进行整合。产生的簇由细胞类型和条件定义,这给下游分析带来了挑战。
# run standard anlaysis workflow
ifnb <- NormalizeData(ifnb)
ifnb <- FindVariableFeatures(ifnb)
ifnb <- ScaleData(ifnb)
ifnb <- RunPCA(ifnb)
ifnb <- FindNeighbors(ifnb, dims = 1:30, reduction = "pca")
ifnb <- FindClusters(ifnb, resolution = 2, cluster.name = "unintegrated_clusters")
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 13999
## Number of edges: 555146
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8153
## Number of communities: 26
## Elapsed time: 23 seconds
ifnb <- RunUMAP(ifnb, dims = 1:30, reduction = "pca", reduction.name = "umap.unintegrated")
DimPlot(ifnb, reduction = "umap.unintegrated", group.by = c("stim", "seurat_clusters"))
整合
我们现在的目标是整合这两种条件的数据,以便来自相同细胞类型/亚群的细胞聚集在一起。
我们经常将此过程称为整合/对齐。当将两个基因组序列比对在一起时,共享/同源区域的识别也可以帮助解释序列之间的差异。同样,对于 scRNA-seq 整合,我们的目标不是消除不同条件下的生物学差异,而是在第一步中了解共享的细胞类型/状态 - 特别是因为这将使我们能够比较这些单个细胞类型的控制刺激和控制概况。
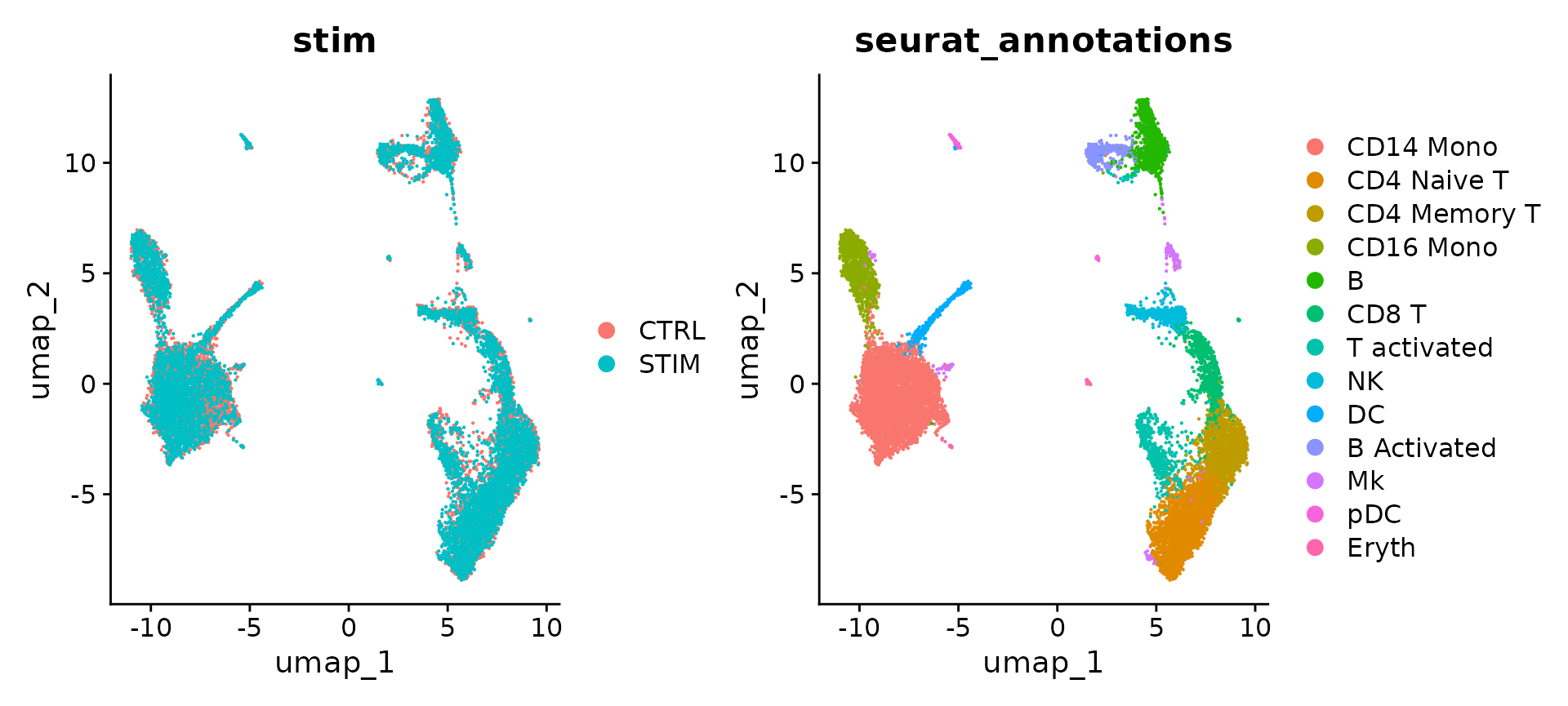
Seurat v5 整合过程旨在返回一个单维缩减,捕获多个层之间共享的方差源,以便处于相似生物状态的细胞能够聚集。该方法返回降维(即integrated.cca),可用于可视化和无监督聚类分析。为了评估性能,我们可以使用 seurat_annotations 元数据列中预加载的单元格类型标签。
ifnb <- IntegrateLayers(object = ifnb, method = CCAIntegration, orig.reduction = "pca", new.reduction = "integrated.cca",
verbose = FALSE)
# re-join layers after integration
ifnb[["RNA"]] <- JoinLayers(ifnb[["RNA"]])
ifnb <- FindNeighbors(ifnb, reduction = "integrated.cca", dims = 1:30)
ifnb <- FindClusters(ifnb, resolution = 1)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 13999
## Number of edges: 588593
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8454
## Number of communities: 17
## Elapsed time: 26 seconds
ifnb <- RunUMAP(ifnb, dims = 1:30, reduction = "integrated.cca")
# Visualization
DimPlot(ifnb, reduction = "umap", group.by = c("stim", "seurat_annotations"))
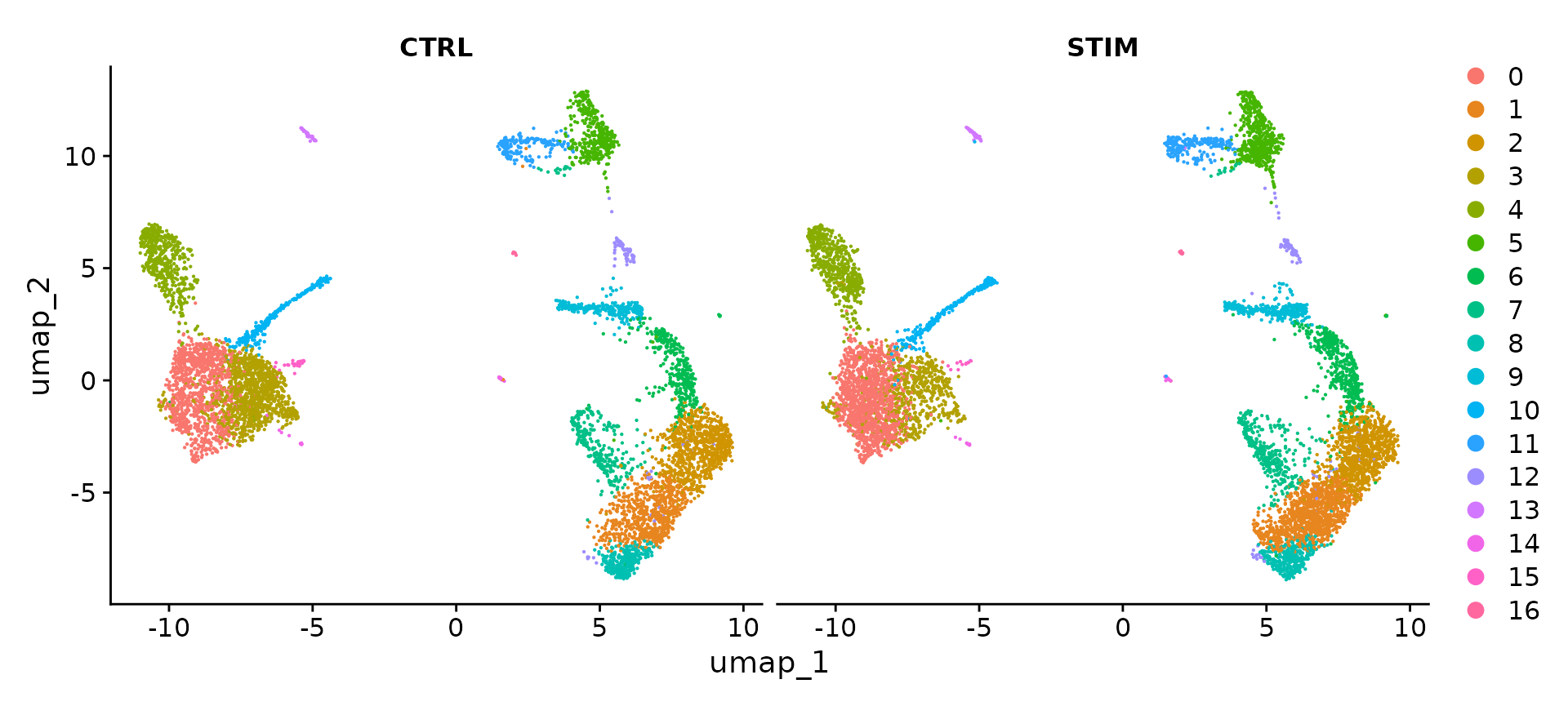
为了并排可视化这两个条件,我们可以使用 split.by 参数来显示按簇着色的每个条件。
DimPlot(ifnb, reduction = "umap", split.by = "stim")
保守细胞类型marker鉴定
为了识别在不同条件下保守的典型细胞类型标记基因,我们提供了 FindConservedMarkers() 函数。该函数对每个数据集/组执行差异基因表达测试,并使用 MetaDE R 包中的荟萃分析方法组合 p 值。例如,无论簇 6(NK 细胞)中的刺激条件如何,我们都可以计算作为保守标记的基因。
Idents(ifnb) <- "seurat_annotations"
nk.markers <- FindConservedMarkers(ifnb, ident.1 = "NK", grouping.var = "stim", verbose = FALSE)
head(nk.markers)
## CTRL_p_val CTRL_avg_log2FC CTRL_pct.1 CTRL_pct.2 CTRL_p_val_adj
## GNLY 0 6.854586 0.943 0.046 0
## NKG7 0 5.358881 0.953 0.085 0
## GZMB 0 5.078135 0.839 0.044 0
## CLIC3 0 5.765314 0.601 0.024 0
## CTSW 0 5.307246 0.537 0.030 0
## KLRD1 0 5.261553 0.507 0.019 0
## STIM_p_val STIM_avg_log2FC STIM_pct.1 STIM_pct.2 STIM_p_val_adj max_pval
## GNLY 0 6.435910 0.956 0.059 0 0
## NKG7 0 4.971397 0.950 0.081 0 0
## GZMB 0 5.151924 0.897 0.060 0 0
## CLIC3 0 5.505208 0.623 0.031 0 0
## CTSW 0 5.240729 0.592 0.035 0 0
## KLRD1 0 4.852457 0.555 0.027 0 0
## minimump_p_val
## GNLY 0
## NKG7 0
## GZMB 0
## CLIC3 0
## CTSW 0
## KLRD1 0
您可以对无监督聚类结果(存储在 seurat_clusters 中)执行相同的分析,并使用这些保守标记来注释数据集中的细胞类型。
带有 split.by 参数的 DotPlot() 函数可用于查看不同条件下的保守细胞类型标记,显示表达水平和簇中表达任何给定基因的细胞百分比。在这里,我们为 14 个簇中的每一个绘制了 2-3 个强标记基因。
# NEEDS TO BE FIXED AND SET ORDER CORRECTLY
Idents(ifnb) <- factor(Idents(ifnb), levels = c("pDC", "Eryth", "Mk", "DC", "CD14 Mono", "CD16 Mono",
"B Activated", "B", "CD8 T", "NK", "T activated", "CD4 Naive T", "CD4 Memory T"))
markers.to.plot <- c("CD3D", "CREM", "HSPH1", "SELL", "GIMAP5", "CACYBP", "GNLY", "NKG7", "CCL5",
"CD8A", "MS4A1", "CD79A", "MIR155HG", "NME1", "FCGR3A", "VMO1", "CCL2", "S100A9", "HLA-DQA1",
"GPR183", "PPBP", "GNG11", "HBA2", "HBB", "TSPAN13", "IL3RA", "IGJ", "PRSS57")
DotPlot(ifnb, features = markers.to.plot, cols = c("blue", "red"), dot.scale = 8, split.by = "stim") +
RotatedAxis()

差异表达基因鉴定
现在我们已经将刺激细胞和对照细胞整合,我们可以开始进行比较分析并观察刺激引起的差异。
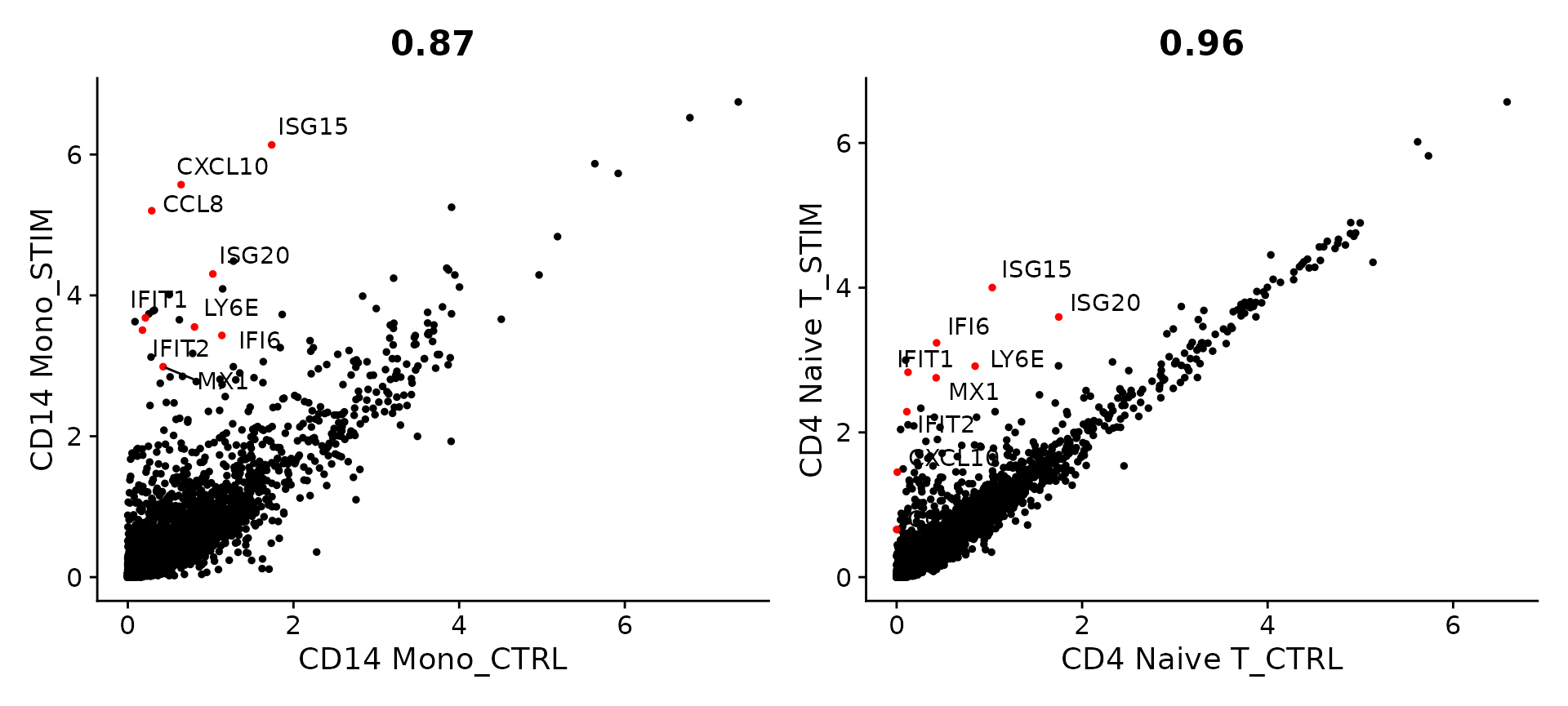
我们可以使用 AggregateExpression 命令将类似类型和条件的细胞聚集在一起,以创建“伪批量”配置文件。作为初步探索性分析,我们可以比较两种细胞类型(初始 CD4 T 细胞和 CD14 单核细胞)的伪批量图谱,并比较刺激前后它们的基因表达谱。我们重点关注对干扰素刺激表现出剧烈反应的基因。正如您所看到的,尽管 CD14 单核细胞表现出更强的转录反应,但许多相同的基因在这两种细胞类型中均上调,并且可能代表保守的干扰素反应途径。
library(ggplot2)
library(cowplot)
theme_set(theme_cowplot())
aggregate_ifnb <- AggregateExpression(ifnb, group.by = c("seurat_annotations", "stim"), return.seurat = TRUE)
genes.to.label = c("ISG15", "LY6E", "IFI6", "ISG20", "MX1", "IFIT2", "IFIT1", "CXCL10", "CCL8")
p1 <- CellScatter(aggregate_ifnb, "CD14 Mono_CTRL", "CD14 Mono_STIM", highlight = genes.to.label)
p2 <- LabelPoints(plot = p1, points = genes.to.label, repel = TRUE)
p3 <- CellScatter(aggregate_ifnb, "CD4 Naive T_CTRL", "CD4 Naive T_STIM", highlight = genes.to.label)
p4 <- LabelPoints(plot = p3, points = genes.to.label, repel = TRUE)
p2 + p4
我们现在可以询问同一类型的细胞在不同条件下哪些基因发生了变化。首先,我们在 meta.data 槽中创建一个列来保存细胞类型和刺激信息,并将当前标识切换到该列。然后我们使用 FindMarkers() 查找刺激 B 细胞和对照 B 细胞之间不同的基因。请注意,此处显示的许多顶级基因与我们之前绘制的核心干扰素反应基因相同。此外,我们看到的 CXCL10 等针对单核细胞和 B 细胞干扰素反应的基因在该列表中也显示出非常重要的意义。
请注意,应谨慎解释从该分析中获得的 p 值,因为这些测试将每个细胞视为独立的重复,并忽略来自同一样本的细胞之间的固有相关性。正如此处所讨论的,跨多种条件的 DE 测试应明确利用多个样本/重复,并且可以在将来自同一样本和亚群的细胞聚集(“伪批量”)在一起后进行。我们在这里不执行此分析,因为数据中有一个重复,但请参阅我们比较健康和糖尿病样本的小插图,作为如何跨条件执行 DE 分析的示例。
ifnb$celltype.stim <- paste(ifnb$seurat_annotations, ifnb$stim, sep = "_")
Idents(ifnb) <- "celltype.stim"
b.interferon.response <- FindMarkers(ifnb, ident.1 = "B_STIM", ident.2 = "B_CTRL", verbose = FALSE)
head(b.interferon.response, n = 15)
## p_val avg_log2FC pct.1 pct.2 p_val_adj
## ISG15 5.387767e-159 5.0588481 0.998 0.233 7.571429e-155
## IFIT3 1.945114e-154 6.1124940 0.965 0.052 2.733468e-150
## IFI6 2.503565e-152 5.4933132 0.965 0.076 3.518260e-148
## ISG20 6.492570e-150 3.0549593 1.000 0.668 9.124009e-146
## IFIT1 1.951022e-139 6.2320388 0.907 0.029 2.741772e-135
## MX1 6.897626e-123 3.9798482 0.905 0.115 9.693234e-119
## LY6E 2.825649e-120 3.7907800 0.898 0.150 3.970885e-116
## TNFSF10 4.007285e-112 6.5802175 0.786 0.020 5.631437e-108
## IFIT2 2.672552e-108 5.5525558 0.786 0.037 3.755738e-104
## B2M 5.283684e-98 0.6104044 1.000 1.000 7.425161e-94
## PLSCR1 4.634658e-96 3.8010721 0.793 0.113 6.513085e-92
## IRF7 2.411149e-94 3.1992949 0.835 0.187 3.388388e-90
## CXCL10 3.708508e-86 8.0906108 0.651 0.010 5.211566e-82
## UBE2L6 5.547472e-83 2.5167981 0.851 0.297 7.795863e-79
## PSMB9 1.716262e-77 1.7715351 0.937 0.568 2.411863e-73
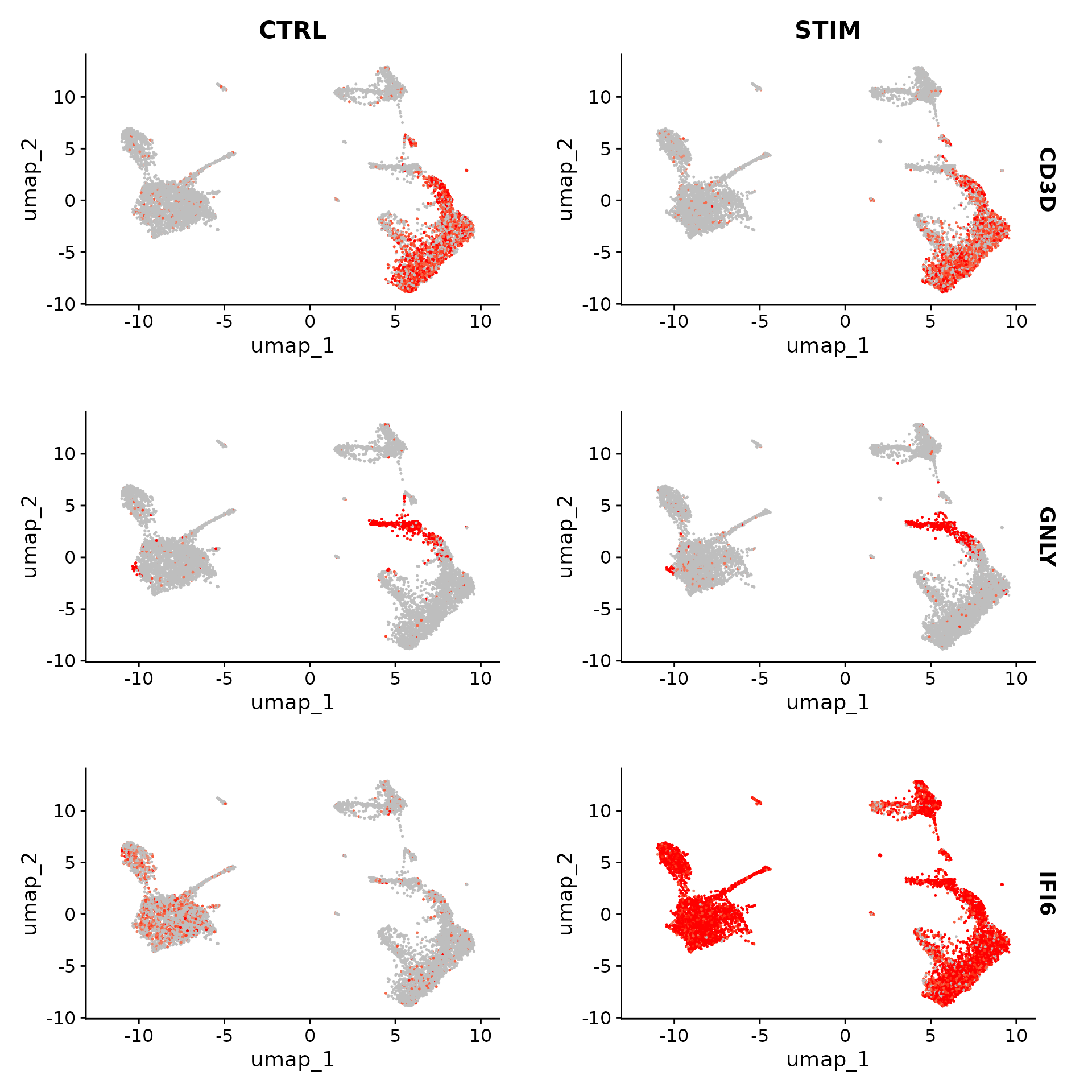
另一种可视化基因表达变化的有用方法是使用 FeaturePlot() 或 VlnPlot() 函数的 split.by 选项。这将显示给定基因列表的特征图,按分组变量(此处为刺激条件)分割。 CD3D 和 GNLY 等基因是典型的细胞类型标记(T 细胞和 NK/CD8 T 细胞),它们几乎不受干扰素刺激的影响,并且在对照组和刺激组中表现出相似的基因表达模式。另一方面,IFI6 和 ISG15 是核心干扰素反应基因,在所有细胞类型中均相应上调。最后,CD14 和 CXCL10 是显示细胞类型特异性干扰素反应的基因。 CD14 单核细胞刺激后 CD14 表达降低,这可能导致监督分析框架中的错误分类,强调了整合分析的价值。干扰素刺激后,CXCL10 在单核细胞和 B 细胞中显示出明显的上调,但在其他细胞类型中则没有。
FeaturePlot(ifnb, features = c("CD3D", "GNLY", "IFI6"), split.by = "stim", max.cutoff = 3, cols = c("grey",
"red"), reduction = "umap")
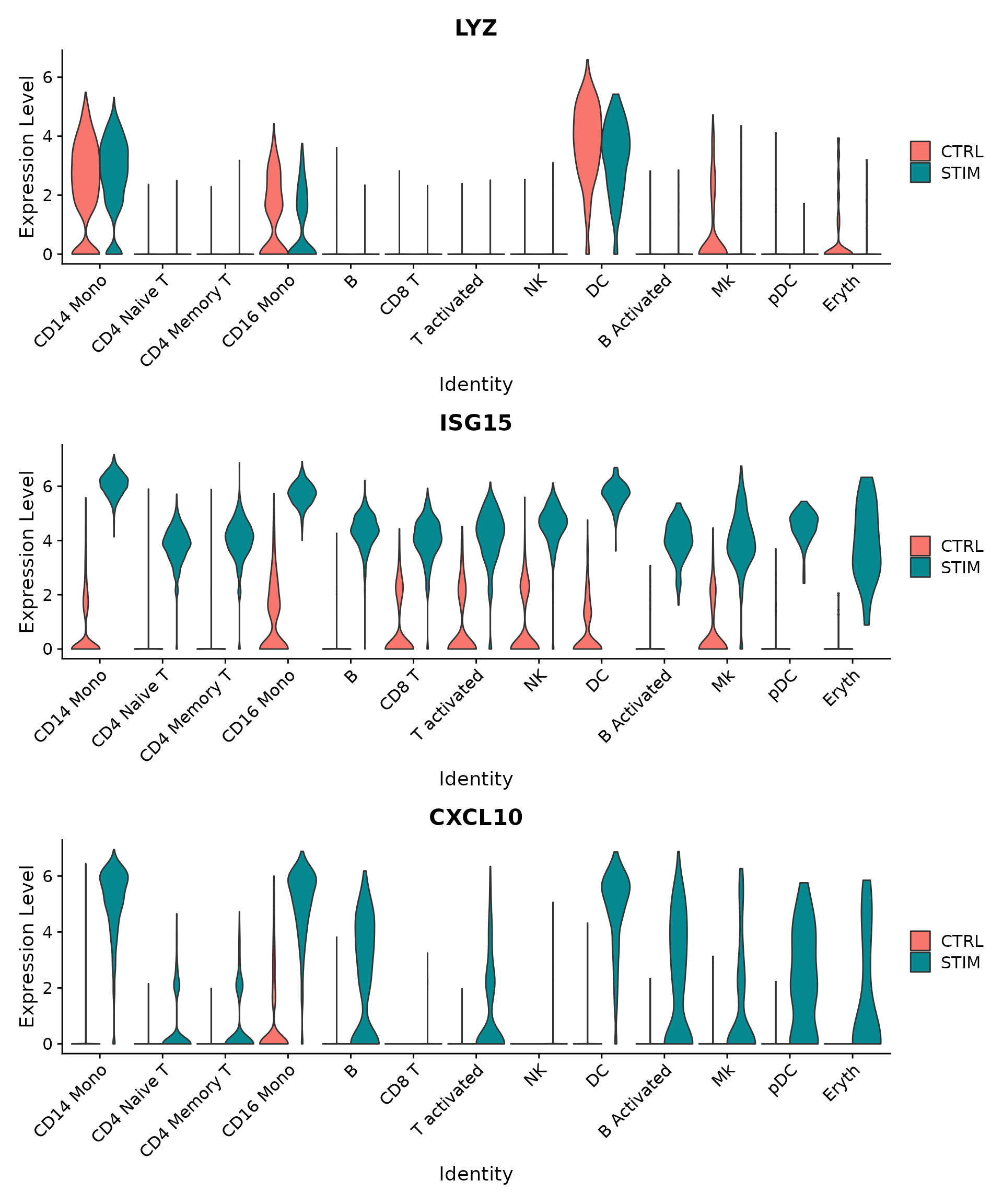
plots <- VlnPlot(ifnb, features = c("LYZ", "ISG15", "CXCL10"), split.by = "stim", group.by = "seurat_annotations",
pt.size = 0, combine = FALSE)
wrap_plots(plots = plots, ncol = 1)
未完待续,欢迎持续关注!








 苏公网安备32011502012024号
苏公网安备32011502012024号