导读
撰写本文的主要目的是:整合 处理与对照后的 PBMC(Human peripheral blood mononuclear cell,人外周血单个核细胞) 数据集以了解细胞类型特异性反应和整合的作用。
本教程介绍了来自 Kang 等人,2017 年 的两组 PBMC 的比对情况。在这个实验中,PBMCs 被分成处理组(刺激组)和对照组,处理组使用 干扰素β 处理。对干扰素的应激导致细胞类型特异性基因表达发生变化,这使得对所有数据的联合分析变得困难。在这里,我们展示了我们的整合策略,如 Stuart 和 Butler 等人,2018 年 所述,执行整合分析以促进常见细胞类型的识别并进行比较分析。虽然此示例只演示了两个数据集(条件)的整合,但这个方法可以扩展到多个数据集。
1. 目的
以下教程旨在为您概述使用 Seurat 整合后对复杂细胞类型进行的各种比较分析。
在这里,我们有以下三个目标:
- 识别两个数据集中都存在的细胞类型
- 获得对照组和处理组中都保守的细胞类型标记(
markers)
- 通过比较数据集来寻找对刺激处理产生特异性反应的细胞类型
2. 创建对象
基因表达矩阵可以在文末的链接找到下载地址,或 点我。我们首先读入两个计数矩阵并创建 Seurat 对象。
# 加载包
library(Seurat)
library(cowplot)
# 读取数据
ctrl.data <- read.table(file = "../data/immune_control_expression_matrix.txt.gz", sep = "\t")
stim.data <- read.table(file = "../data/immune_stimulated_expression_matrix.txt.gz", sep = "\t")
# 创建 对照组 对象
ctrl <- CreateSeuratObject(counts = ctrl.data, project = "IMMUNE_CTRL", min.cells = 5)
ctrl$stim <- "CTRL"
ctrl <- subset(ctrl, subset = nFeature_RNA > 500)
ctrl <- NormalizeData(ctrl, verbose = FALSE)
ctrl <- FindVariableFeatures(ctrl, selection.method = "vst", nfeatures = 2000)
# 创建 处理组 对象
stim <- CreateSeuratObject(counts = stim.data, project = "IMMUNE_STIM", min.cells = 5)
stim$stim <- "STIM"
stim <- subset(stim, subset = nFeature_RNA > 500)
stim <- NormalizeData(stim, verbose = FALSE)
stim <- FindVariableFeatures(stim, selection.method = "vst", nfeatures = 2000)
3. 整合
然后,我们使用 FindIntegrationAnchors 函数识别 anchors(锚点),该函数将 Seurat 对象列表作为输入,并使用这些锚点,利用 IntegrateData 函数将两个数据集整合在一起。
# 识别 anchors
immune.anchors <- FindIntegrationAnchors(object.list = list(ctrl, stim), dims = 1:20)
# 整合
immune.combined <- IntegrateData(anchorset = immune.anchors, dims = 1:20)
4. 整体分析
现在我们可以对所有细胞进行一个综合分析!
DefaultAssay(immune.combined) <- "integrated"
# 运行可视化和聚类的标准工作流程
immune.combined <- ScaleData(immune.combined, verbose = FALSE)
immune.combined <- RunPCA(immune.combined, npcs = 30, verbose = FALSE)
# t-SNE and 聚类
immune.combined <- RunUMAP(immune.combined, reduction = "pca", dims = 1:20)
immune.combined <- FindNeighbors(immune.combined, reduction = "pca", dims = 1:20)
immune.combined <- FindClusters(immune.combined, resolution = 0.5)
# 可视化
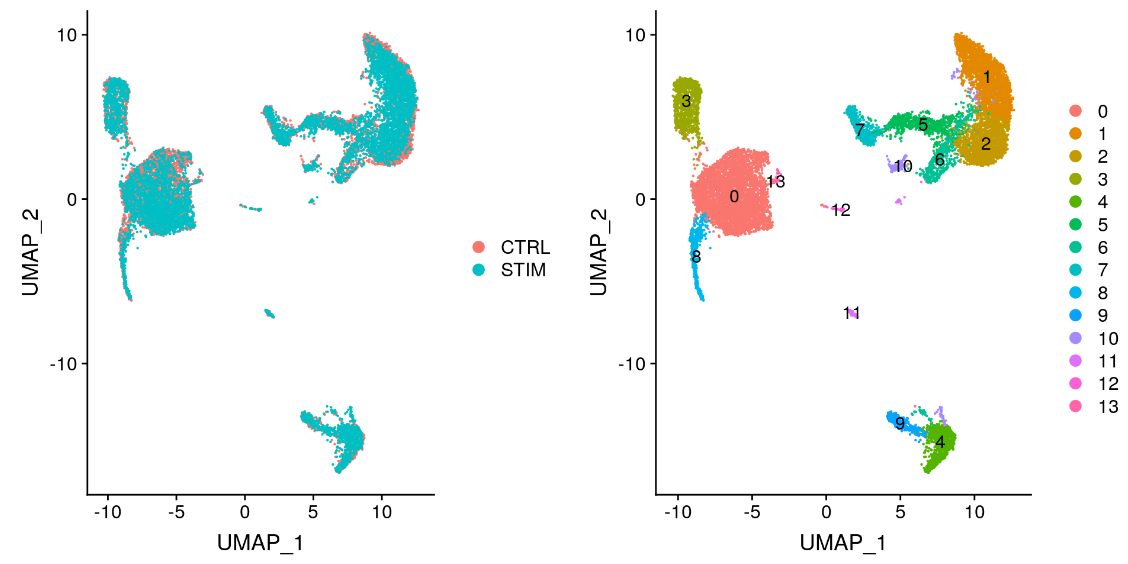
p1 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")
p2 <- DimPlot(immune.combined, reduction = "umap", label = TRUE)
plot_grid(p1, p2)
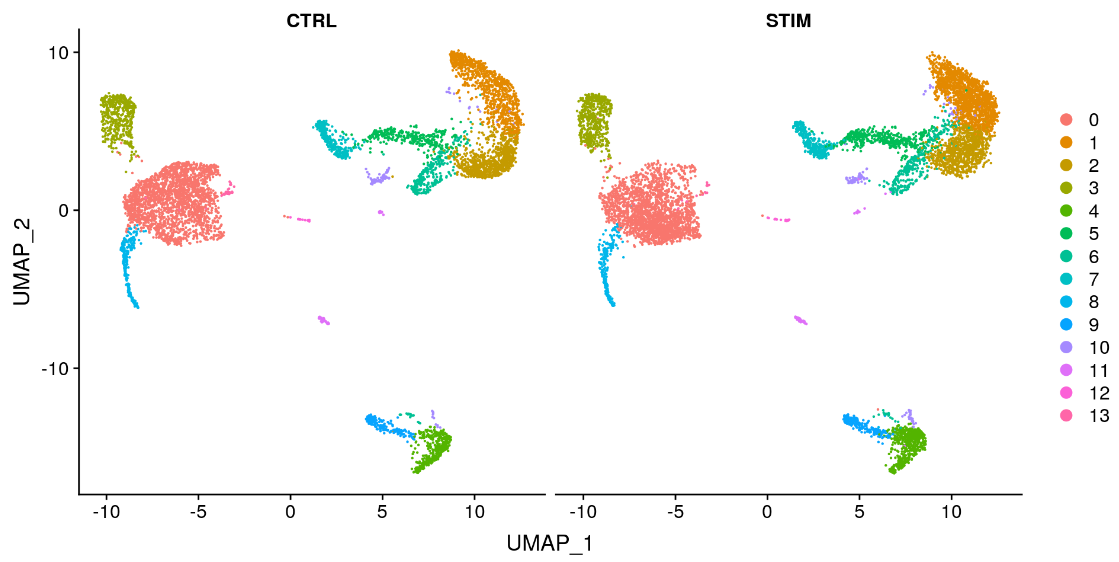
为了并排可视化这两个条件,我们可以使用 split.by 参数来显示按簇着色的每个条件。
DimPlot(immune.combined, reduction = "umap", split.by = "stim")
5. 保守marker鉴定
为了识别跨条件保守的细胞类型标记基因,Seurat 提供了 FindConservedMarkers 函数。此函数对每个数据集或组执行差异基因表达 test,并使用 MetaDE R 包中的元分析(Meta-analysis)方法组合 p 值。例如,我们可以计算簇 7(NK 细胞)中无论条件如何,都是保守的标记基因。
DefaultAssay(immune.combined) <- "RNA"
nk.markers <- FindConservedMarkers(immune.combined, ident.1 = 7, grouping.var = "stim", verbose = FALSE)
head(nk.markers)

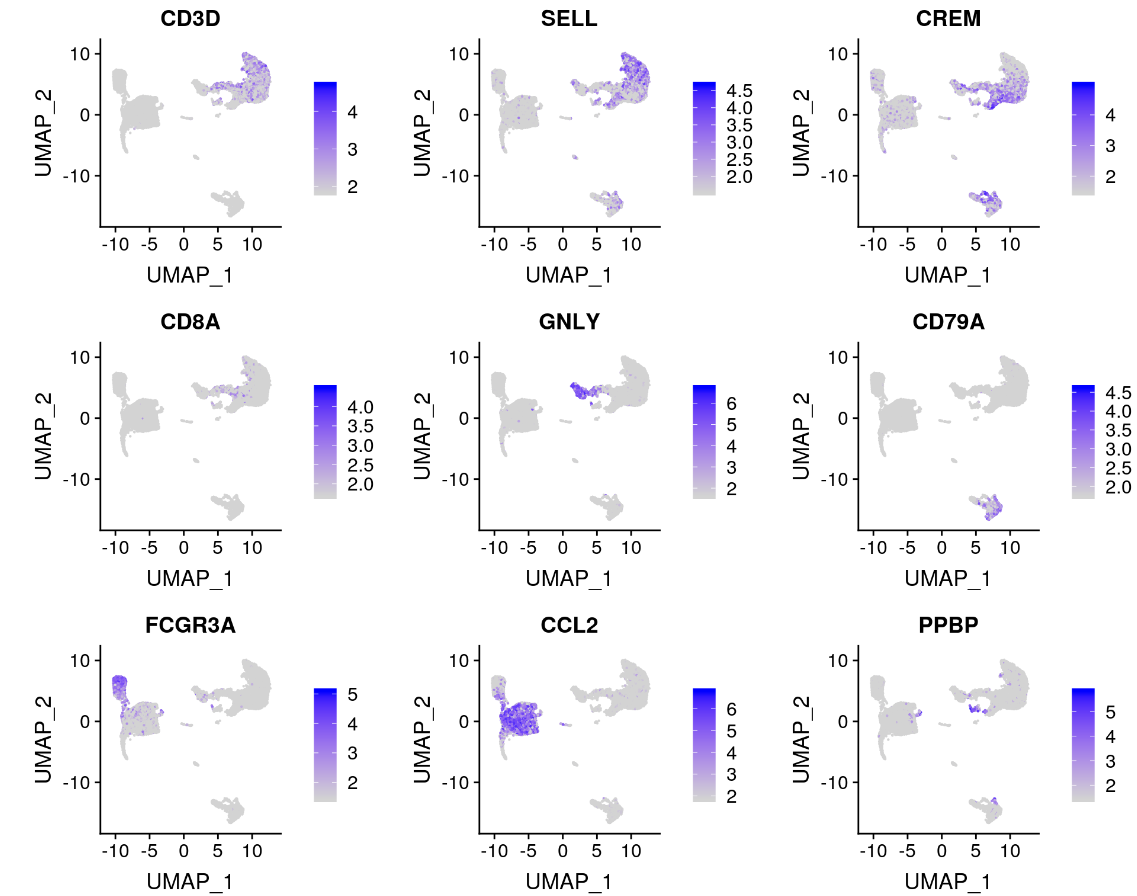
我们可以探索每个簇的这些标记基因,并使用它们,将我们的簇注释为特定的细胞类型。
FeaturePlot(immune.combined, features = c("CD3D", "SELL", "CREM", "CD8A", "GNLY", "CD79A", "FCGR3A",
"CCL2", "PPBP"), min.cutoff = "q9")
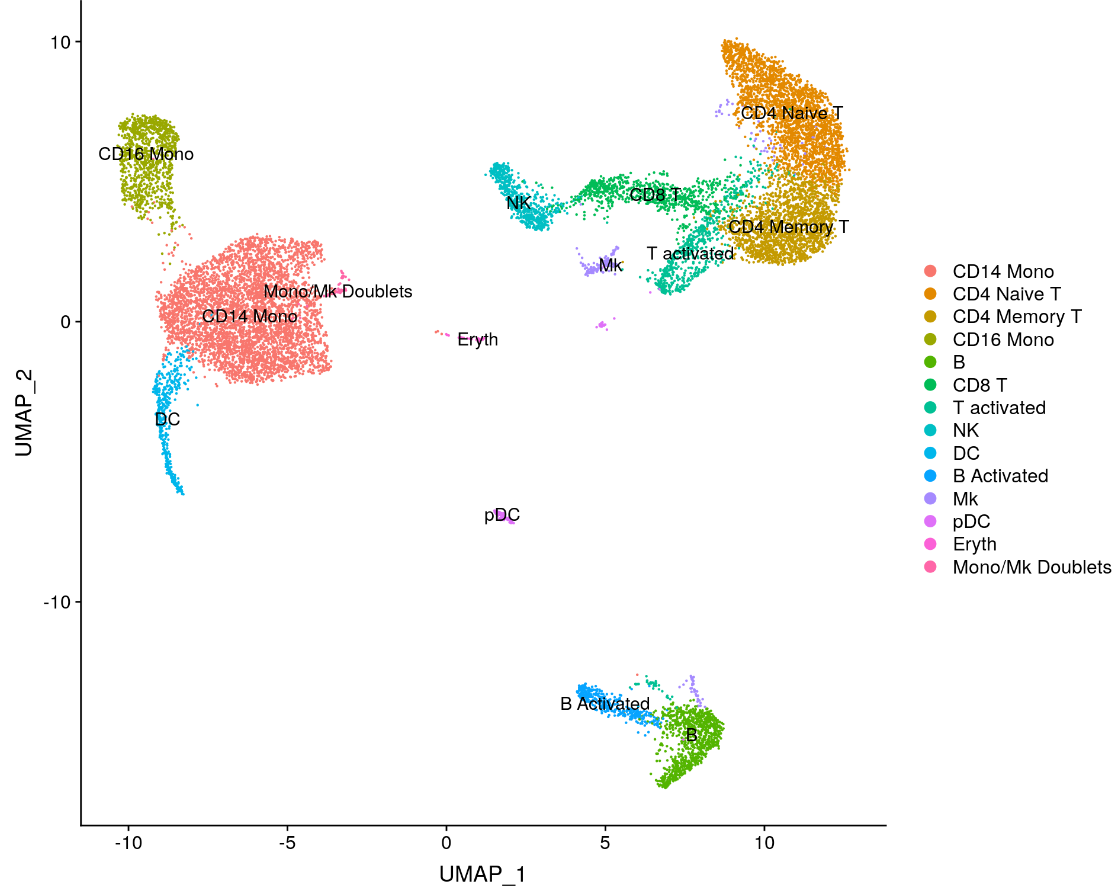
immune.combined <- RenameIdents(immune.combined, `0` = "CD14 Mono", `1` = "CD4 Naive T", `2` = "CD4 Memory T",
`3` = "CD16 Mono", `4` = "B", `5` = "CD8 T", `6` = "T activated", `7` = "NK", `8` = "DC", `9` = "B Activated",
`10` = "Mk", `11` = "pDC", `12` = "Eryth", `13` = "Mono/Mk Doublets")
DimPlot(immune.combined, label = TRUE)
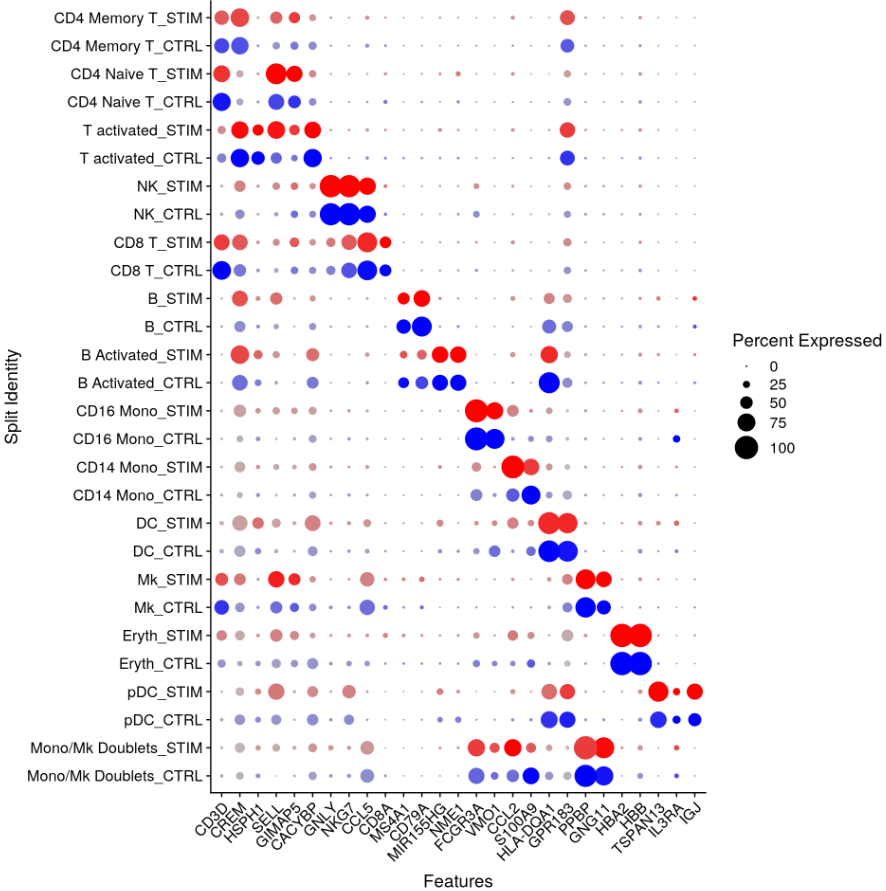
带有 split.by 参数的 DotPlot 函数可用于查看跨条件的保守细胞类型标记,显示表达水平和表达任何给定基因的簇中细胞的百分比。在这里,我们为 13 个聚类中的每一个绘制了 2-3 个强标记基因。
Idents(immune.combined) <- factor(Idents(immune.combined), levels = c("Mono/Mk Doublets", "pDC",
"Eryth", "Mk", "DC", "CD14 Mono", "CD16 Mono", "B Activated", "B", "CD8 T", "NK", "T activated",
"CD4 Naive T", "CD4 Memory T"))
markers.to.plot <- c("CD3D", "CREM", "HSPH1", "SELL", "GIMAP5", "CACYBP", "GNLY", "NKG7", "CCL5",
"CD8A", "MS4A1", "CD79A", "MIR155HG", "NME1", "FCGR3A", "VMO1", "CCL2", "S100A9", "HLA-DQA1",
"GPR183", "PPBP", "GNG11", "HBA2", "HBB", "TSPAN13", "IL3RA", "IGJ")
DotPlot(immune.combined, features = rev(markers.to.plot), cols = c("blue", "red"), dot.scale = 8,
split.by = "stim") + RotatedAxis()
6. 跨条件识别差异表达基因
现在我们已经对齐了受刺激组(处理组)细胞和对照组细胞,我们可以开始进行比较分析并查看刺激引起的差异。观察这些变化的一种方法是绘制受刺激细胞和对照细胞的平均表达,并在散点图上寻找异常值的基因。在这里,我们取受刺激和对照 naive T 细胞和 CD14 单核细胞群的平均表达,并生成散点图,突出显示对干扰素刺激有显着反应的基因。
t.cells <- subset(immune.combined, idents = "CD4 Naive T")
Idents(t.cells) <- "stim"
avg.t.cells <- log1p(AverageExpression(t.cells, verbose = FALSE)$RNA)
avg.t.cells$gene <- rownames(avg.t.cells)
cd14.mono <- subset(immune.combined, idents = "CD14 Mono")
Idents(cd14.mono) <- "stim"
avg.cd14.mono <- log1p(AverageExpression(cd14.mono, verbose = FALSE)$RNA)
avg.cd14.mono$gene <- rownames(avg.cd14.mono)
genes.to.label = c("ISG15", "LY6E", "IFI6", "ISG20", "MX1", "IFIT2", "IFIT1", "CXCL10", "CCL8")
p1 <- ggplot(avg.t.cells, aes(CTRL, STIM)) + geom_point() + ggtitle("CD4 Naive T Cells")
p1 <- LabelPoints(plot = p1, points = genes.to.label, repel = TRUE)
p2 <- ggplot(avg.cd14.mono, aes(CTRL, STIM)) + geom_point() + ggtitle("CD14 Monocytes")
p2 <- LabelPoints(plot = p2, points = genes.to.label, repel = TRUE)
plot_grid(p1, p2)
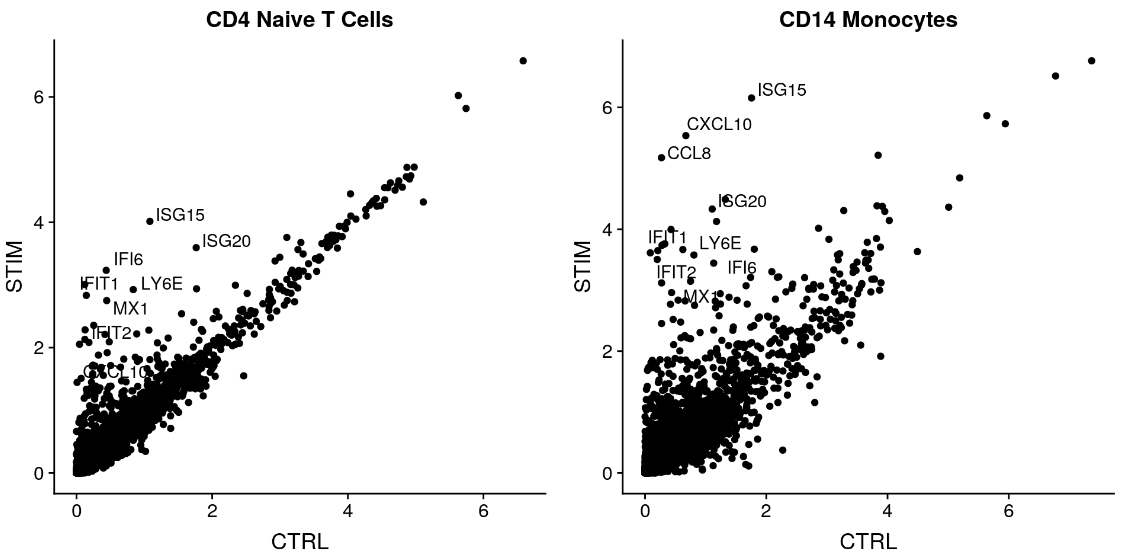
正如您所看到的,许多相同的基因在这两种细胞类型中都上调,并且可能代表了一种保守的干扰素反应途径。
因为我们有信心在不同条件下识别出常见的细胞类型,所以我们可以查看相同类型细胞在不同条件下哪些基因会发生变化。首先,我们在 meta.data 中创建一个列来保存细胞类型和处理信息,并将当前标识切换到该列。然后使用 FindMarkers 来查找受刺激 B 细胞和对照 B 细胞之间不同的基因。请注意,此处显示的许多 Top 基因与我们之前绘制的核心干扰素反应基因相同。此外,我们看到的 CXCL10 等特定于单核细胞和 B 细胞干扰素反应的基因在此列表中也显示出非常重要的意义。
immune.combined$celltype.stim <- paste(Idents(immune.combined), immune.combined$stim, sep = "_")
immune.combined$celltype <- Idents(immune.combined)
Idents(immune.combined) <- "celltype.stim"
b.interferon.response <- FindMarkers(immune.combined, ident.1 = "B_STIM", ident.2 = "B_CTRL", verbose = FALSE)
head(b.interferon.response, n = 15)

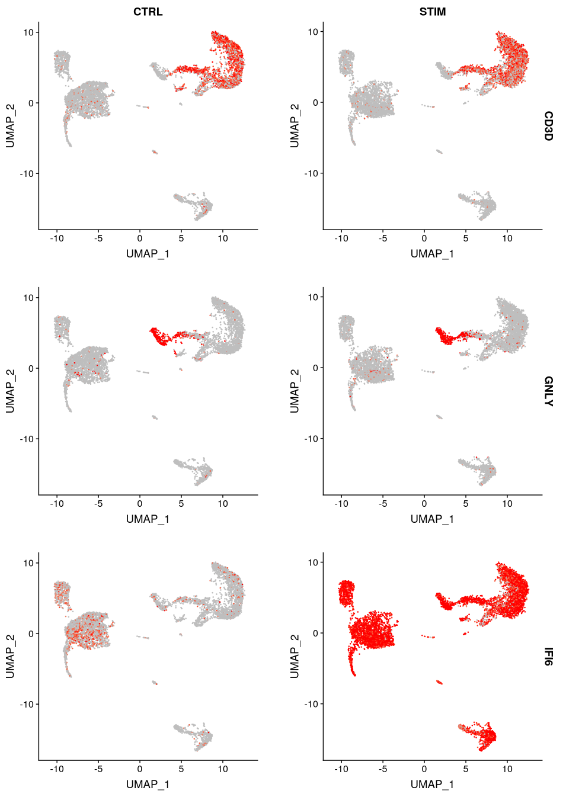
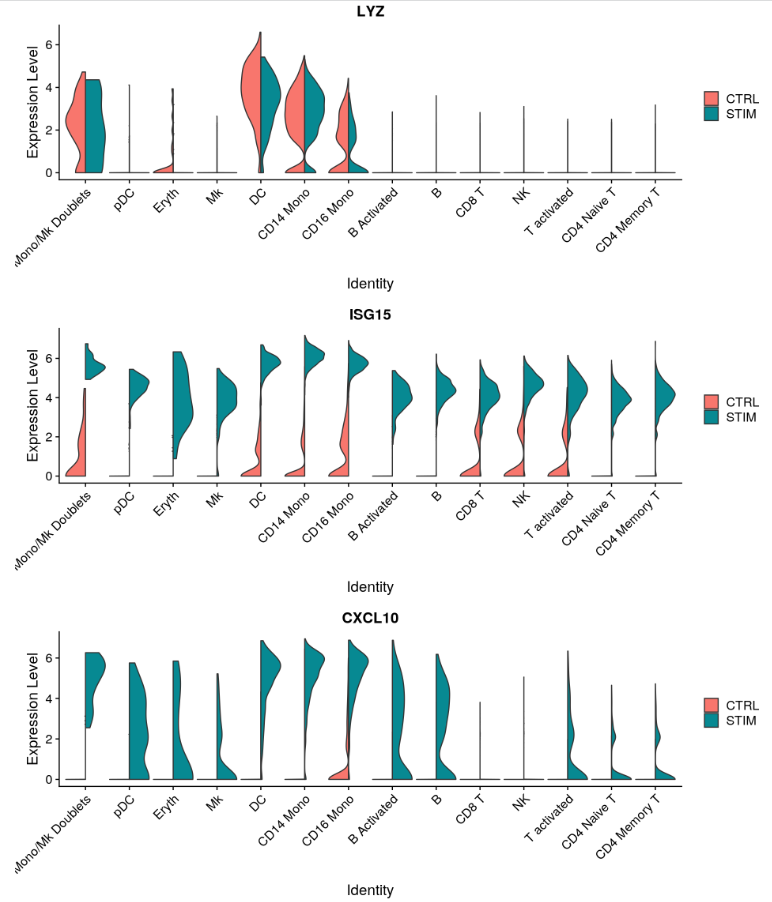
另一种可视化基因表达变化的方法是使用 FeaturePlot 或 VlnPlot 函数的 split.by 选项。这将显示给定基因列表的特征图,按分组变量(此处为刺激条件)拆分。CD3D 和 GNLY 等基因是典型的细胞类型标记(用于 T 细胞和 NK/CD8 T 细胞),它们几乎不受干扰素刺激的影响,并且在对照组和受刺激组中显示出相似的基因表达模式。另一方面,IFI6 和 ISG15 是核心干扰素反应基因,并在所有细胞类型中上调。最后,CD14 和 CXCL10 是显示细胞类型特异性干扰素反应的基因。刺激 CD14 单核细胞后 CD14 表达降低,这可能导致监督分析框架中发生错误分类,强调了整合分析的价值。CXCL10 在干扰素刺激后在单核细胞和 B 细胞中显示出明显的上调,但在其他细胞类型中则没有。
FeaturePlot(immune.combined, features = c("CD3D", "GNLY", "IFI6"), split.by = "stim", max.cutoff = 3,
cols = c("grey", "red"))
plots <- VlnPlot(immune.combined, features = c("LYZ", "ISG15", "CXCL10"), split.by = "stim", group.by = "celltype",
pt.size = 0, combine = FALSE)
CombinePlots(plots = plots, ncol = 1)








 苏公网安备32011502012024号
苏公网安备32011502012024号