reference映射简介
在本文中,我们首先构建一个reference,然后演示如何利用该reference来注释新的查询数据集。生成后,该reference可用于通过cell类型标签传输和将查询cell投影到reference UMAP 等任务来分析其他查询数据集。值得注意的是,这不需要纠正底层原始查询数据,因此如果有高质量的reference可用,这可能是一种有效的策略。
数据集预处理
出于本示例的目的,我们选择了通过四种技术生成的人类胰岛细胞数据集:CelSeq (GSE81076) CelSeq2 (GSE85241)、Fluidigm C1 (GSE86469) 和 SMART-Seq2 (E-MTAB-5061)。为了方便起见,我们通过 SeuratData 包分发此数据集。元数据包含四个数据集中每个细胞的技术(技术列)和细胞类型注释(细胞类型列)。
library(Seurat)
library(SeuratData)
library(ggplot2)
InstallData("panc8")
作为演示,我们将使用各种技术来构建参考。然后,我们将剩余的数据集映射到该参考上。我们首先从四种技术中选择cell,并在不进行整合的情况下进行分析。
panc8 <- LoadData("panc8")
table(panc8$tech)
##
## celseq celseq2 fluidigmc1 indrop smartseq2
## 1004 2285 638 8569 2394
# we will use data from 2 technologies for the reference
pancreas.ref <- subset(panc8, tech %in% c("celseq2", "smartseq2"))
pancreas.ref[["RNA"]] <- split(pancreas.ref[["RNA"]], f = pancreas.ref$tech)
# pre-process dataset (without integration)
pancreas.ref <- NormalizeData(pancreas.ref)
pancreas.ref <- FindVariableFeatures(pancreas.ref)
pancreas.ref <- ScaleData(pancreas.ref)
pancreas.ref <- RunPCA(pancreas.ref)
pancreas.ref <- FindNeighbors(pancreas.ref, dims = 1:30)
pancreas.ref <- FindClusters(pancreas.ref)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 4679
## Number of edges: 174953
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.9180
## Number of communities: 19
## Elapsed time: 0 seconds
pancreas.ref <- RunUMAP(pancreas.ref, dims = 1:30)
DimPlot(pancreas.ref, group.by = c("celltytpe", "tech"))
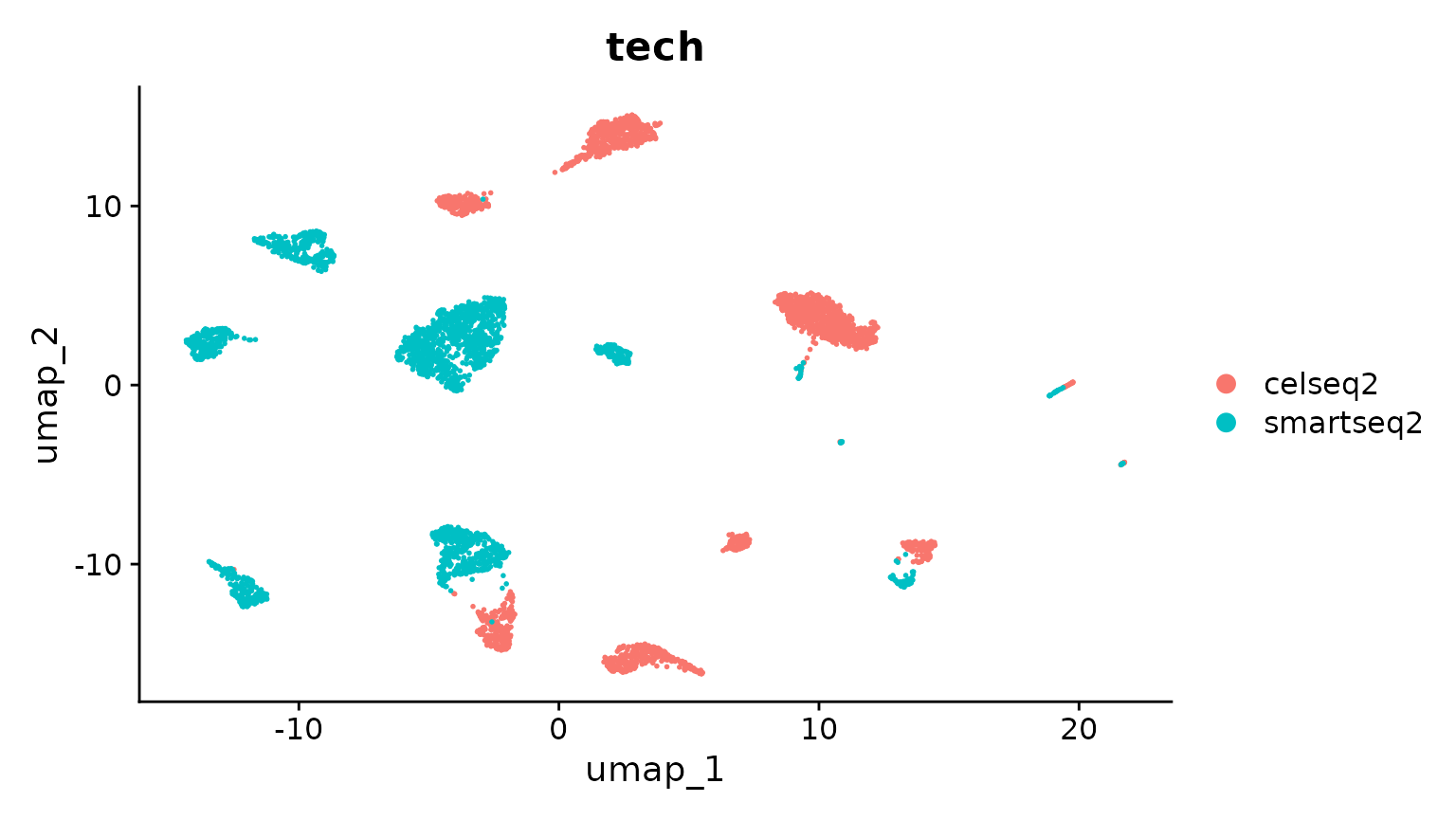
接下来,我们将数据集整合到reference中。
pancreas.ref <- IntegrateLayers(object = pancreas.ref, method = CCAIntegration, orig.reduction = "pca",
new.reduction = "integrated.cca", verbose = FALSE)
pancreas.ref <- FindNeighbors(pancreas.ref, reduction = "integrated.cca", dims = 1:30)
pancreas.ref <- FindClusters(pancreas.ref)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 4679
## Number of edges: 190152
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8680
## Number of communities: 15
## Elapsed time: 0 seconds
pancreas.ref <- RunUMAP(pancreas.ref, reduction = "integrated.cca", dims = 1:30)
DimPlot(pancreas.ref, group.by = c("tech", "celltype"))
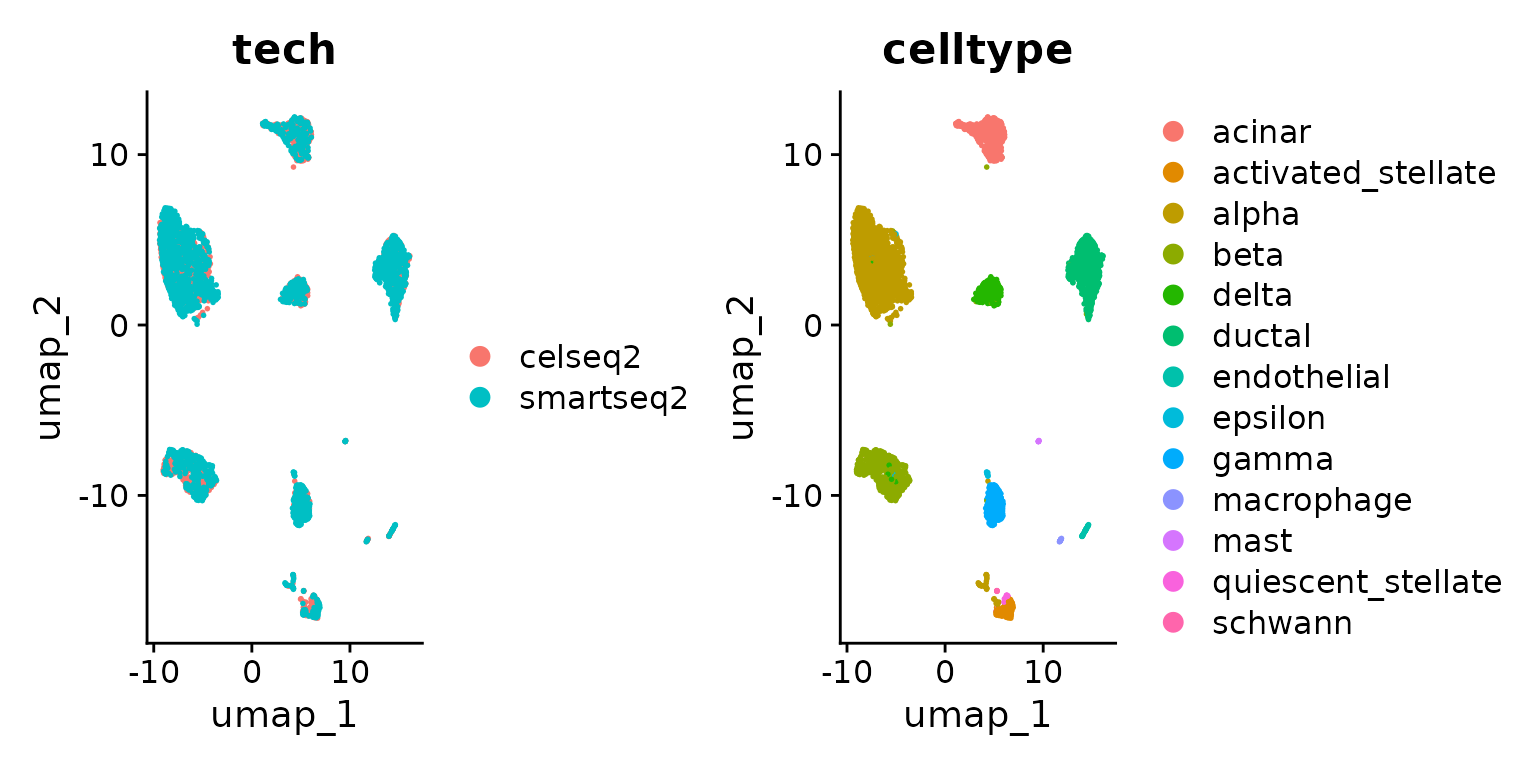
使用整合后的reference进行细胞类型分类
Seurat 还支持将参考数据(或元数据)投影到查询对象上。虽然许多方法都是保守的(两个过程都从识别锚点开始),但数据传输和集成之间有两个重要区别:
- 在数据传输中,Seurat 不会更正或修改查询表达式数据。
- 在数据传输中,Seurat 有一个选项(默认设置)将引用的 PCA 结构投影到查询上,而不是使用 CCA 学习联合结构。我们通常建议在 scRNA-seq 数据集之间投影数据时使用此选项。
找到锚点后,我们使用 TransferData() 函数根据参考数据(参考单元类型标签的向量)对查询cell进行分类。 TransferData() 返回一个包含预测 ID 和预测分数的矩阵,我们可以将其添加到查询元数据中。
# select two technologies for the query datasets
pancreas.query <- subset(panc8, tech %in% c("fluidigmc1", "celseq"))
pancreas.query <- NormalizeData(pancreas.query)
pancreas.anchors <- FindTransferAnchors(reference = pancreas.ref, query = pancreas.query, dims = 1:30,
reference.reduction = "pca")
predictions <- TransferData(anchorset = pancreas.anchors, refdata = pancreas.ref$celltype, dims = 1:30)
pancreas.query <- AddMetaData(pancreas.query, metadata = predictions)
因为我们拥有来自完整整合分析的原始标签注释,所以我们可以评估预测的细胞类型注释与完整参考的匹配程度。
在此示例中,我们发现细胞类型分类具有很高的一致性,超过 96% 的细胞被正确标记。
pancreas.query$prediction.match <- pancreas.query$predicted.id == pancreas.query$celltype
table(pancreas.query$prediction.match)
##
## FALSE TRUE
## 63 1579
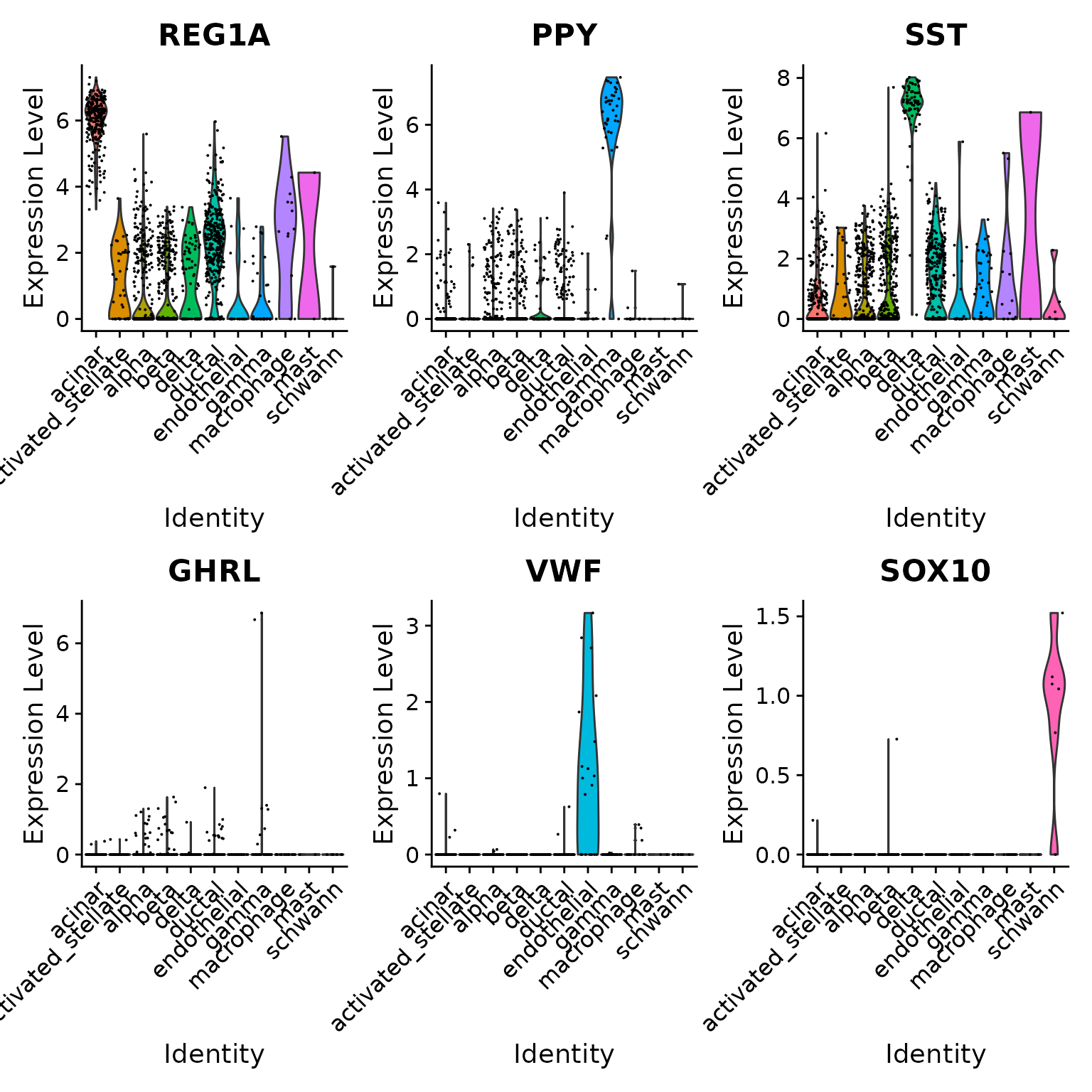
为了进一步验证这一点,我们可以检查特定胰岛细胞群的一些典型细胞类型标记。请注意,即使其中一些细胞类型仅由一个或两个细胞(例如 epsilon 细胞)代表,我们仍然能够正确对它们进行分类。
table(pancreas.query$predicted.id)
##
## acinar activated_stellate alpha beta
## 262 39 436 419
## delta ductal endothelial gamma
## 73 330 19 41
## macrophage mast schwann
## 15 2 6
VlnPlot(pancreas.query, c("REG1A", "PPY", "SST", "GHRL", "VWF", "SOX10"), group.by = "predicted.id")
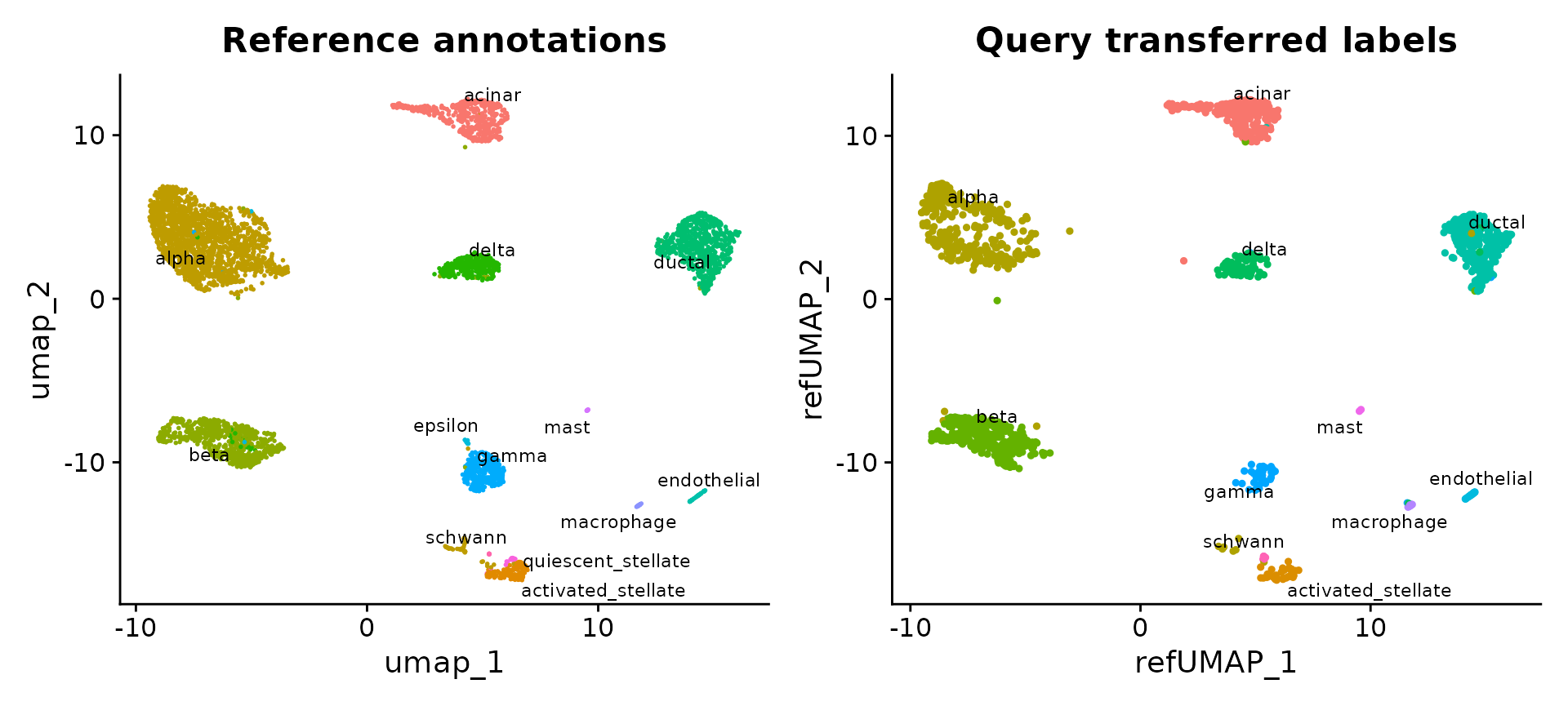
UMAP 投影
我们还支持将查询投影到参考 UMAP 结构上。这可以通过计算参考 UMAP 模型然后调用 MapQuery() 而不是 TransferData() 来实现。
pancreas.ref <- RunUMAP(pancreas.ref, dims = 1:30, reduction = "integrated.cca", return.model = TRUE)
pancreas.query <- MapQuery(anchorset = pancreas.anchors, reference = pancreas.ref, query = pancreas.query,
refdata = list(celltype = "celltype"), reference.reduction = "pca", reduction.model = "umap")
p1 <- DimPlot(pancreas.ref, reduction = "umap", group.by = "celltype", label = TRUE, label.size = 3,
repel = TRUE) + NoLegend() + ggtitle("Reference annotations")
p2 <- DimPlot(pancreas.query, reduction = "ref.umap", group.by = "predicted.celltype", label = TRUE,
label.size = 3, repel = TRUE) + NoLegend() + ggtitle("Query transferred labels")
p1 + p2








 苏公网安备32011502012024号
苏公网安备32011502012024号