数据集
在本文中,我们将对利用Slide-seq v2技术获得的小鼠海马区数据集进行深入分析。
为了方便数据获取,您可以利用我们的SeuratData包,具体操作示例如下。一旦安装了该数据集,您只需输入 ?ssHippo,即可查看构建Seurat对象时所使用的命令列表。
InstallData("ssHippo")
slide.seq <- LoadData("ssHippo")
预处理
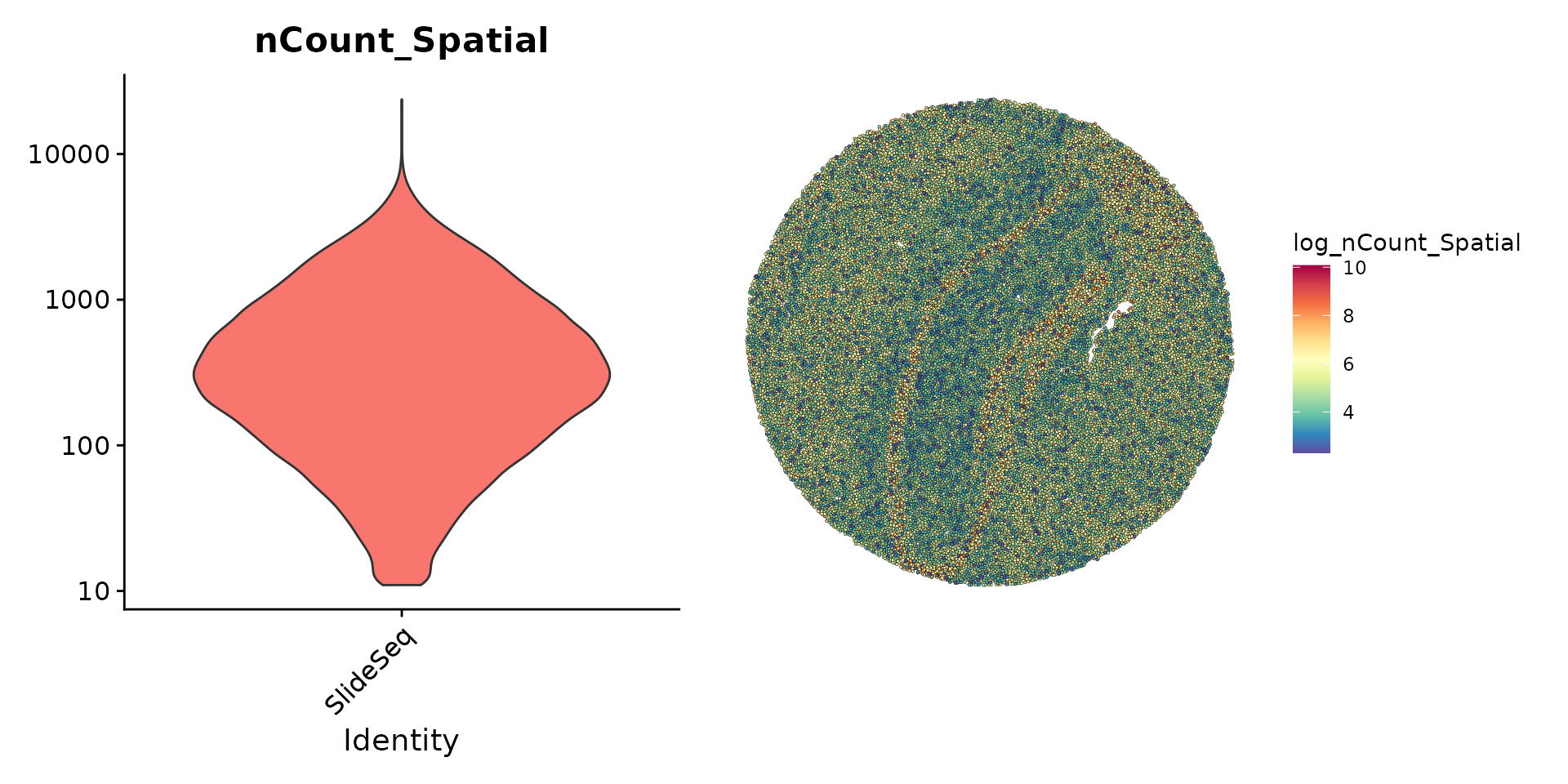
在处理珠子的基因表达数据的初步预处理过程中,我们采用了与其他空间Seurat分析以及常规的单细胞RNA测序实验相似的方法。我们发现,尽管许多珠子的UMI计数非常低,但我们决定保留所有已检测到的珠子,以便进行后续的分析工作。
plot1 <- VlnPlot(slide.seq, features = "nCount_Spatial", pt.size = 0, log = TRUE) + NoLegend()
slide.seq$log_nCount_Spatial <- log(slide.seq$nCount_Spatial)
plot2 <- SpatialFeaturePlot(slide.seq, features = "log_nCount_Spatial") + theme(legend.position = "right")
wrap_plots(plot1, plot2)
然后,我们使用 sctransform 对数据进行标准化,并执行标准的 scRNA-seq 降维和聚类工作流程。
slide.seq <- SCTransform(slide.seq, assay = "Spatial", ncells = 3000, verbose = FALSE)
slide.seq <- RunPCA(slide.seq)
slide.seq <- RunUMAP(slide.seq, dims = 1:30)
slide.seq <- FindNeighbors(slide.seq, dims = 1:30)
slide.seq <- FindClusters(slide.seq, resolution = 0.3, verbose = FALSE)
然后,我们可以在 UMAP 空间(使用 DimPlot())或使用 SpatialDimPlot() 在珠坐标空间中可视化聚类结果。
plot1 <- DimPlot(slide.seq, reduction = "umap", label = TRUE)
plot2 <- SpatialDimPlot(slide.seq, stroke = 0)
plot1 + plot2

SpatialDimPlot(slide.seq, cells.highlight = CellsByIdentities(object = slide.seq, idents = c(1,
6, 13)), facet.highlight = TRUE)

与 scRNA-seq 整合
为了方便对Slide-seq数据集进行细胞类型的标注,我们正在使用Saunders和Macosko等人在2018年发表的一个现有小鼠单细胞RNA-seq海马区数据集。
ref <- readRDS("/brahms/shared/vignette-data/mouse_hippocampus_reference.rds")
ref <- UpdateSeuratObject(ref)
文章中提供的原始注释可以在Seurat对象的细胞元数据中找到。这些注释覆盖了多个“分辨率”级别,包括从宽泛的类别(ref$class)到细胞类型内部的子群组(ref$subcluster)。我们将基于对细胞类型注释(ref$celltype)的一次调整来进行工作,我们认为这样的调整在保持平衡方面做得很好。
我们将首先执行Seurat的标签转移方法,以此来预测每个珠子的主要细胞类型。
anchors <- FindTransferAnchors(reference = ref, query = slide.seq, normalization.method = "SCT",
npcs = 50)
predictions.assay <- TransferData(anchorset = anchors, refdata = ref$celltype, prediction.assay = TRUE,
weight.reduction = slide.seq[["pca"]], dims = 1:50)
slide.seq[["predictions"]] <- predictions.assay
然后我们可以可视化一些主要预期类别的预测分数。
DefaultAssay(slide.seq) <- "predictions"
SpatialFeaturePlot(slide.seq, features = c("Dentate Principal cells", "CA3 Principal cells", "Entorhinal cortex",
"Endothelial tip", "Ependymal", "Oligodendrocyte"), alpha = c(0.1, 1))

slide.seq$predicted.id <- GetTransferPredictions(slide.seq)
Idents(slide.seq) <- "predicted.id"
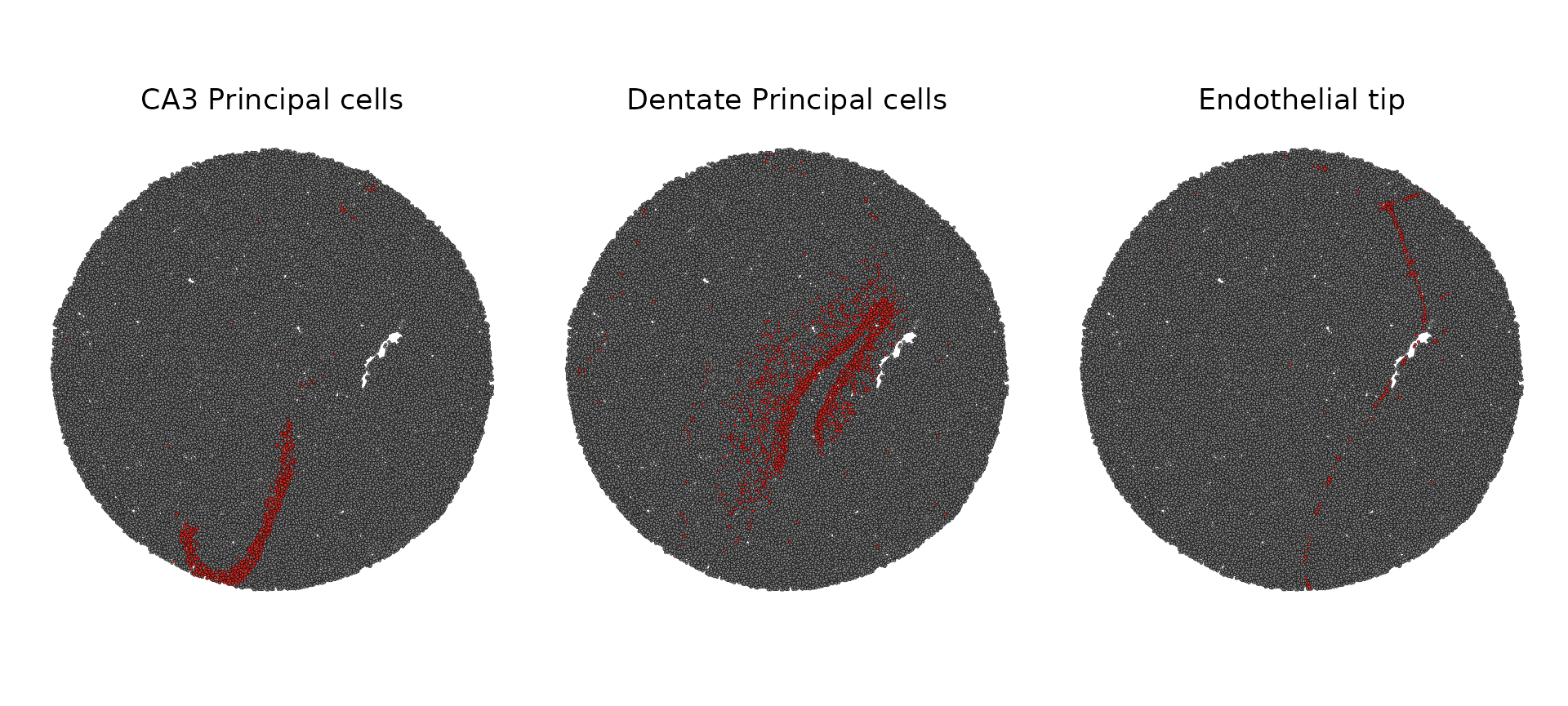
SpatialDimPlot(slide.seq, cells.highlight = CellsByIdentities(object = slide.seq, idents = c("CA3 Principal cells",
"Dentate Principal cells", "Endothelial tip")), facet.highlight = TRUE)








 苏公网安备32011502012024号
苏公网安备32011502012024号