单细胞 RNA 测序(scRNA-seq)基础知识可查看以下文章:
单细胞 RNA 测序(scRNA-seq)工作流程入门
单细胞 RNA 测序(scRNA-seq)细胞分离与扩增
单细胞 RNA 测序(scRNA-seq)SRA 数据下载及 fastq-dumq 数据拆分
单细胞 RNA 测序(scRNA-seq)Cellranger 流程入门和数据质控
单细胞RNA测序(scRNA-seq)构建人类参考基因组注释
细胞定量是scRNA-seq重要的分析步骤,主要是进行细胞与基因的定量, cell ranger将比对、质控、定量都封装了起来,使用起来也相当便捷。
1. 参考基因组和注释文件准备
1.1 参考基因组、注释下载和参考基因组索引构建
参考基因组、注释下载注释和参考基因组索引参考以下文章:
单细胞RNA测序(scRNA-seq)构建人类参考基因组注释
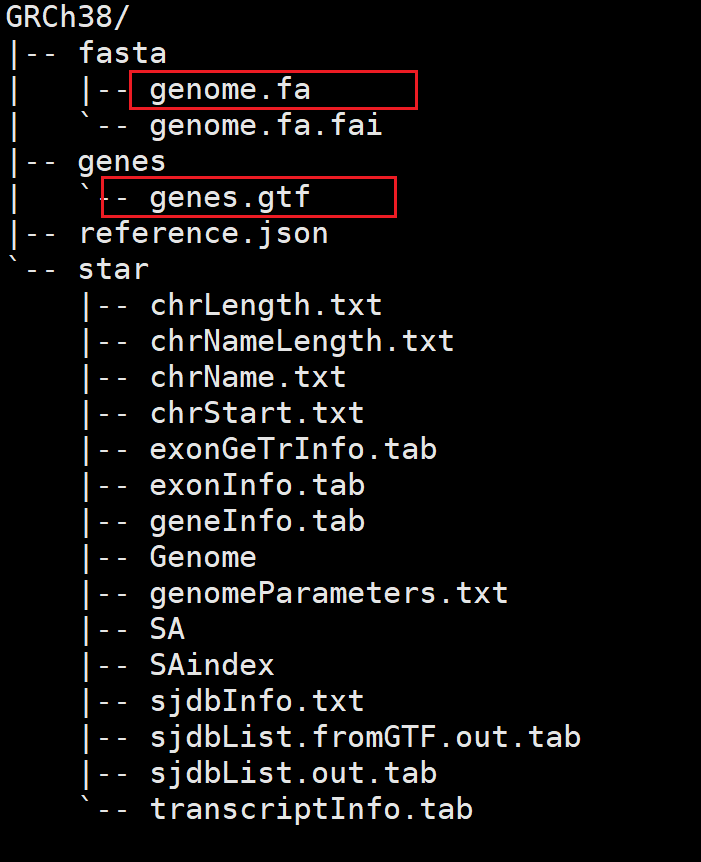
构建完成后,GRCh38( cellranger mkref --genome 参数内容为指定输出文件夹)目录会出现以下内容。
# genes中gtf文件为.gz格式则解压
gzip -d genes.gtf.gz
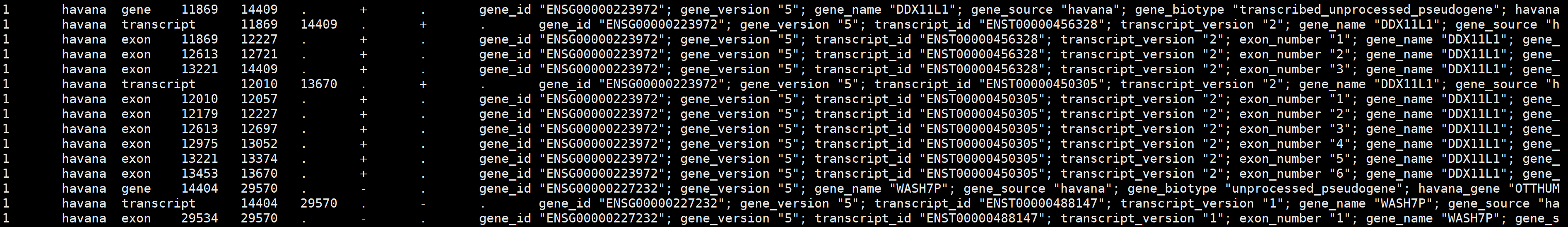
# 查看genes/genes.gtf信息
grep -v '^#' genes.gtf |less -S
1.2 注释基因信息更新
- 在fasta/genome.fa的fasta文件基础上增加序列信息;
- 在genes/genes.gtf的gtf文件基础上增加注释信息(根据GTF文件格式);
- 重新执行运行cellranger mkref流程
2 cellranger count细胞定量
2.1 测序数据fastq文件命名说明
# 一般命名方式
[Sample Name]_S1_L00[Lane Number]_[Read Type]_001.fastq.gz
# 其中Read Type有以下三种:
# I1: Sample index read (optional)
# R1: Read 1
# R2: Read 2
# 查看rawdata目录下fastq.gz文件
ls -lh rawdata
rawdata目录文件列表:

2.2 Shell脚本执行单个样本的细胞定量
cellranger count流程采用**STAT作为比对工具,其具有比对速度快和灵敏度高**等特点。
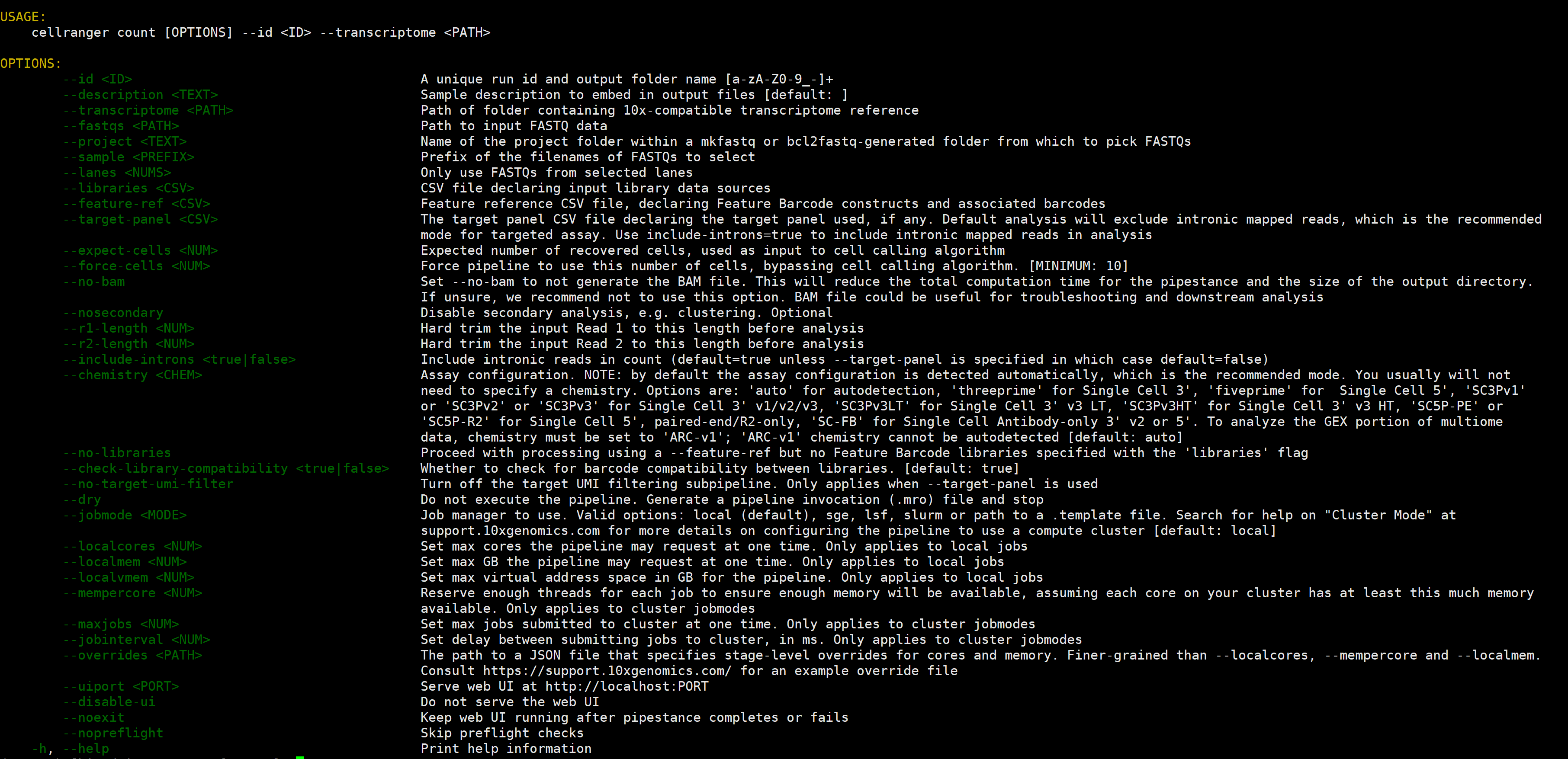
# 查看count用法
cellranger count -h
sh sample_count.sh 执行脚本,进行单个样本的细胞定量。
# id: 输出文件存放的目录名称
id=SRR7692286
# transcriptome: 与CellRanger兼容的参考基因组目录
transcriptome=reference/GRCh38
# fastqs: 存储mkfastq或自定义的测序文件目录
fastqs=rawdata目录
# sample: 与fastq文件[Sample Name],即前缀
sample=SRR7692286
# 细胞数量,与实验设计一致
export_cells=1000
cellranger count --id=$id \
--transcriptome=$transcriptome \
--fastqs=$fastqs \
--sample=$sample \
--expect-cells=$export_cells \
--nosecondary
参数--nosecondary表示只生成表达矩阵文件,不执行后续的降维、聚类和可视化等分析步骤。
出现以下信息说明运行中:
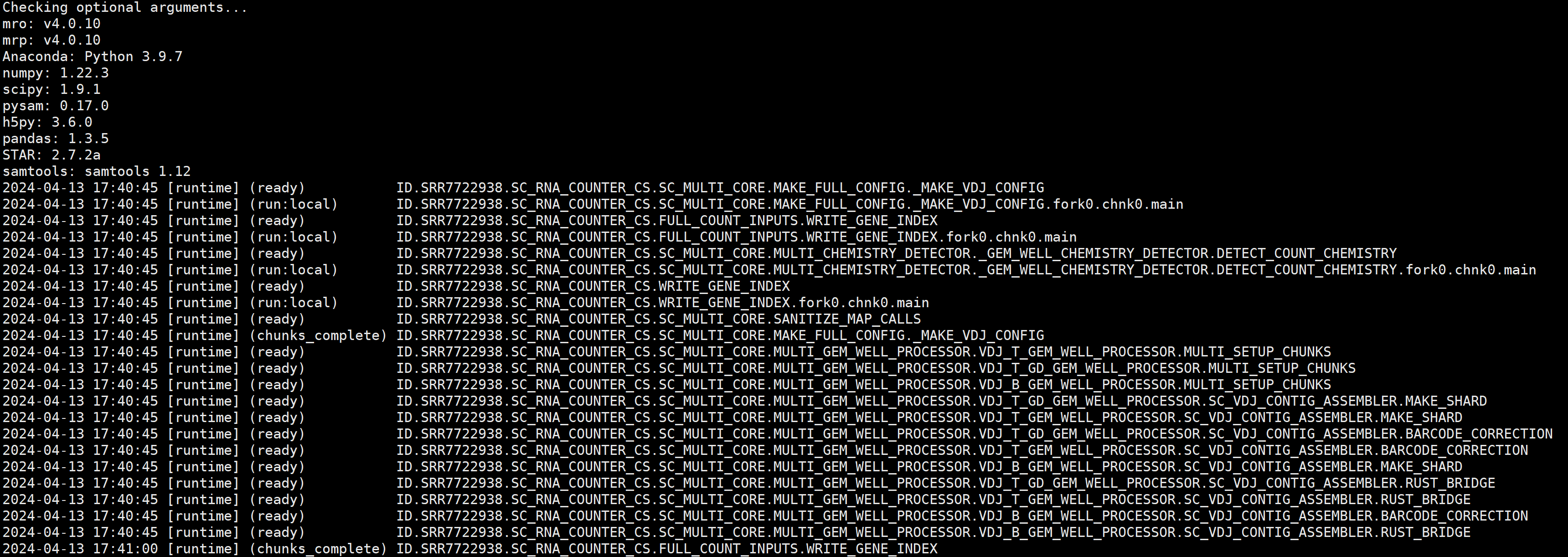
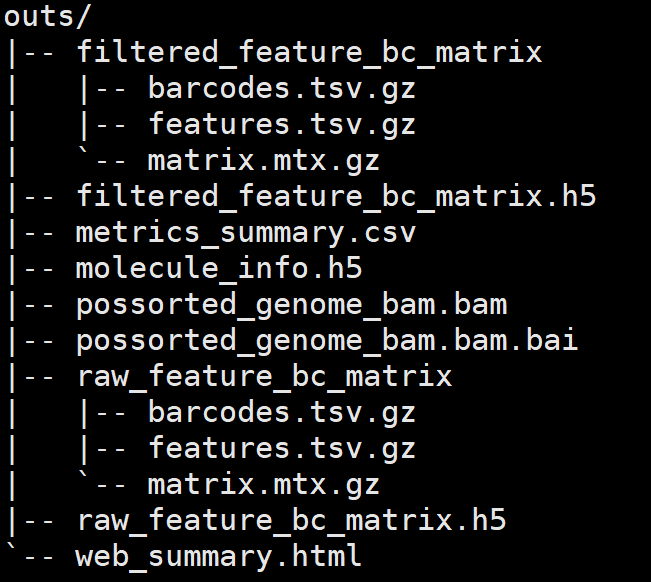
单个样本结果目录如下:
结果目录及文件说明见下文。

2.3 Shell脚本批量执行样本的细胞定量
nohup sh all_count.sh > all_count.log 2>&1 & 后台执行脚本,花费时间较长。
# sh all_count.sh
# 遍历文件,批量执行细胞定量
cat SRR_Acc_List.txt|while read srr; do
# 跳过上一步测试定量单个样本的编号
# if [ "$srr" == "SRR7692286" ];then
# continue
# fi
# id: 输出文件存放的目录名称
id=$srr
# transcriptome: 与CellRanger兼容的参考基因组目录
transcriptome=reference/GRCh38
# fastqs: 存储mkfastq或自定义的测序文件目录
fastqs=rawdata
# sample: 与fastq文件[Sample Name],即前缀
sample=$srr
# 细胞数量,与实验设计一致
export_cells=1000
echo "running cell count: $id"
cellranger count --id=$id \
--transcriptome=$transcriptome \
--fastqs=$fastqs \
--sample=$sample \
--expect-cells=$export_cells \
--nosecondary
done
# 查询分析日志进度
# cat all_count.log
3. 比对结果的分析
比对完之后,利用GTF文件将比对的reads溯源回外显子区、内含子区、基因间区,使用MAPQ值(mapping quality)可以判断比对的可信度,值越大越可信,MAPQ=255时认为确定。
如果一条read的50%以上与外显子有交集,那么就认为reads在外显区;如果不在外显子区,与内含子有交集,那么就认为在内含子区; 与外显子、内含子都没有交集,那么认为在基因间区。
如果上面的外显子区域reads同时比对上有注释转录本上的外显子,并且在同一条链上,那么认为这个reads也比对到了转录组
如果只比对到单个基因的注释信息,那么认为reads是特异比对到转录组的(uniquely /confidently mapped ),此类的reads才会拿来做接下来的UMI 计数。
4. 细胞定量结果文件说明
web_summary.html:官方说明 summary HTML 文件
metrics_summary.csv:包含数据摘要信息
possorted_genome_bam.bam:比对bam文件
possorted_genome_bam.bam.bai:bam索引文件
filtered_gene_bc_matrices(重要结果目录):是重要的一个目录,下面又包含了 barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz,作为下游Seurat、Scater、Monocle等分析的输入文件.
filtered_feature_bc_matrix.h5:过滤掉的barcode信息HDF5格式文件
raw_feature_bc_matrix:原始barcode信息
raw_feature_bc_matrix.h5:原始barcode信息的HDF5 格式文件
analysis: 数据分析目录,包含聚类clustering(有graph-based & k-means)、差异分析diffexp、主成分线性降维分析pca、非线性降维tsne
molecule_info.h5:下一步进行aggregate使用的文件
cloupe.cloupe:官方的可视化工具Loupe Cell Browser 输入文件
5. 多个细胞定量结果的整合
当处理多个生物学样本或者一个样本存在多个重复/文库时,最好是先分别对每个文库进行单独的count定量,然后将定量结果使用cellranger aggr整合起来。
# 获取样本molecule_info.h5文件相对路径
find SRR7692286 -type f -name molecule_info.h5
# SRR7692286/outs/molecule_info.h5
5.1 Shell脚本批量整合多个细胞定量结果
cellranger aggr支持csv文件格式如下:
We support one of the following modes:
- If you are looking to aggr outputs from count by specifying the molecule_info.h5, the following headers are required: '**sample_id,molecule_h5**'
- If you are looking to aggr outputs from vdj by specifying the vdj_contig_info.pb, the following headers are required: 'sample_id,vdj_contig_info,donor,origin'
- If you are looking to aggr outputs from multi by specifying the output folder, the following headers are required: 'sample_id,sample_outs'
sh aggr.sh
执行脚本进行整合。
# 创建count文件夹
mkdir count
# 文件已存在则删除
if [ -f "aggr.csv" ];then
rm aggr.csv
fi
# 添加sample_id,molecule_h5列名
echo "sample_id,molecule_h5" >> aggr.csv
# 循环查找
cat SRR_Acc_List.txt|while read srr; do
# 将样本SRR目录移动至count目录下
mv ${srr} ./count
# 查找样本目录下molecule_info.h5文件路径
h5_path=`find count/${srr} -type f -name molecule_info.h5`
# ,号分割追加至aggr.csv文件中
echo "${srr},${h5_path}" >> aggr.csv
done
# 输出结果目录已存在则删除
if [ -d "SRR_AGGR" ];then
rm -rf SRR_AGGR
fi
# 结果输出到SRR_AGGR目录中
cellranger aggr --id=SRR_AGGR --csv=aggr.csv --normalize=mapped
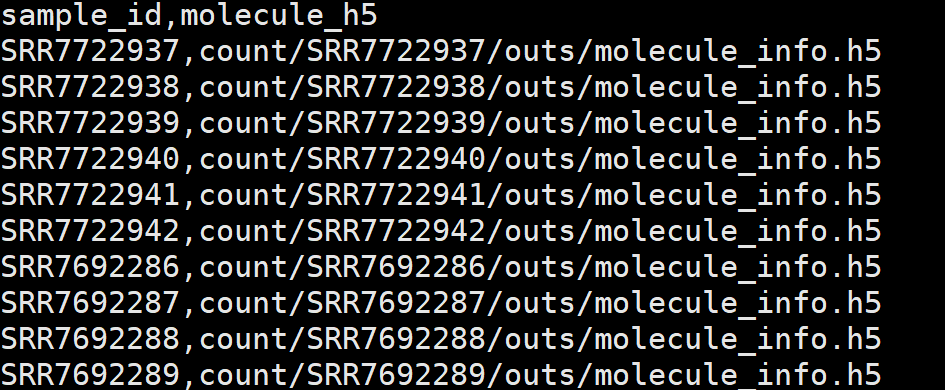
aggr.csv 文件内容如下:






 苏公网安备32011502012024号
苏公网安备32011502012024号