单细胞 RNA 测序入门可以查看以下文章:
单细胞 RNA 测序(scRNA-seq)工作流程入门
单细胞RNA测序(scRNA-seq)细胞分离与扩增
prefetch 软件安装可参考以下文章:
prefetch软件安装
1. NCBI查询scRNA-seq SRA数据
NCBI SRA数据地址: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA484204&o=acc_s%3Aa
点击Accession List下载包含SRR编号信息的文本文件 - SRR_Acc_List.txt。 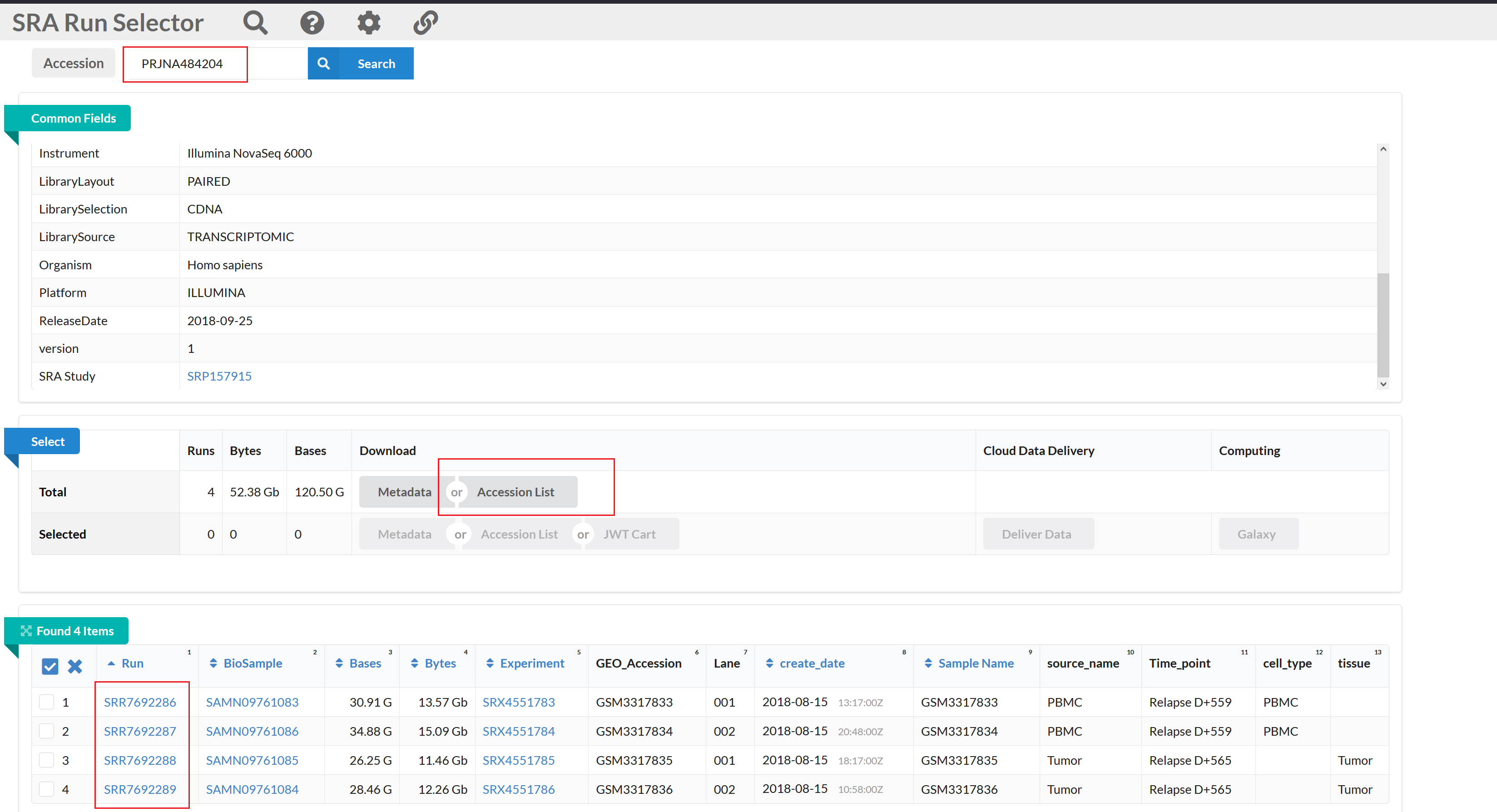
SRR_Acc_List.txt文件内容:

2. 批量下载SRA数据
使用--split-files来替代--split-3 ,就可以生成3个文件。第1个文件的所有序列都是8bp,第2个文件26bp,第3个文件91bp,判断第3个文件时包含测序reads的文件。
######## 单个SRR数据下载与拆分(测试) ########
prefetch SRR7692286
# 后台下载
# nohup prefetch SRR7692286 &
# fastq-dump为-A为指定文件名, --gzip为输出.gz压缩文件
fastq-dump --gzip --split-files -A SRR7692286 SRR7692286.sra
# 拆分sra文件, fastq-dump拆分报错,可尝试使用fasterq-dump
# fasterq-dump --split-files -A SRR7692286 SRR7692286.sra
######## 批量SRR数据下载与拆分 ########
# 根据SRR_Acc_List.txt批量下载,nohup为后台下载
prefetch --option-file SRR_Acc_List.txt
# 后台下载
# nohup prefetch --option-file SRR_Acc_List.txt &
# 批量拆分sra文件为fastq.gz
cat SRR_Acc_List.txt|while read srr; do (fastq-dump --gzip --split-files -A $srr ${srr}.sra); done
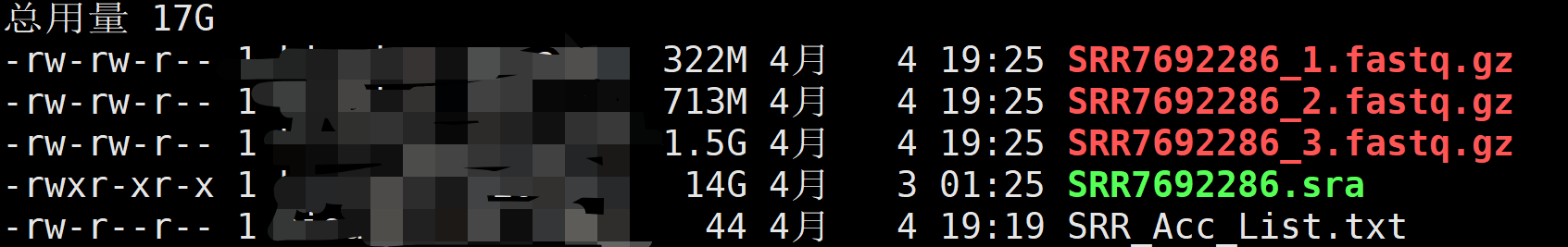
3. fastq-dumq拆分SRA为fastq文件
10X单细胞数据相对比较复杂,其测序文库中包括index、barcode、UMI和测序reads。因此需要对SRA文件进行拆分以获取上述文件,拆分需要使用fastq-dump软件,为sra-tool工具中的软件之一。
# conda安装
conda install -c bioconda sra-tools
4. 了解10X文库组成
R1: 26 表示10X barcode 的 16bp碱基 + 10bp UMI
i7: 8表示 8bp 样本index序列
Read 2: 98 中星号符号表示长度不固定
4.1 i7 sample index的作用?
i7 sample index(library barcode)是加到Illumina测序接头上的,保证多个测序文库可以在同一个flow-cell上或者同一个lane上进行混合测序(multiplexed)。不同的项目index不同,但在96孔板的每个孔中都加入了4种不同的index oligos混合,其作用就是在CellRanger mkfastq 功能(BCL转fastq)中体现出来的,它自动识别样本index名称(例如:SA-GA-A1),将具有相同4种oligo的fq文件组合在一起表示同一个样本,从而保证了一个测序lane上可以容纳多个样本。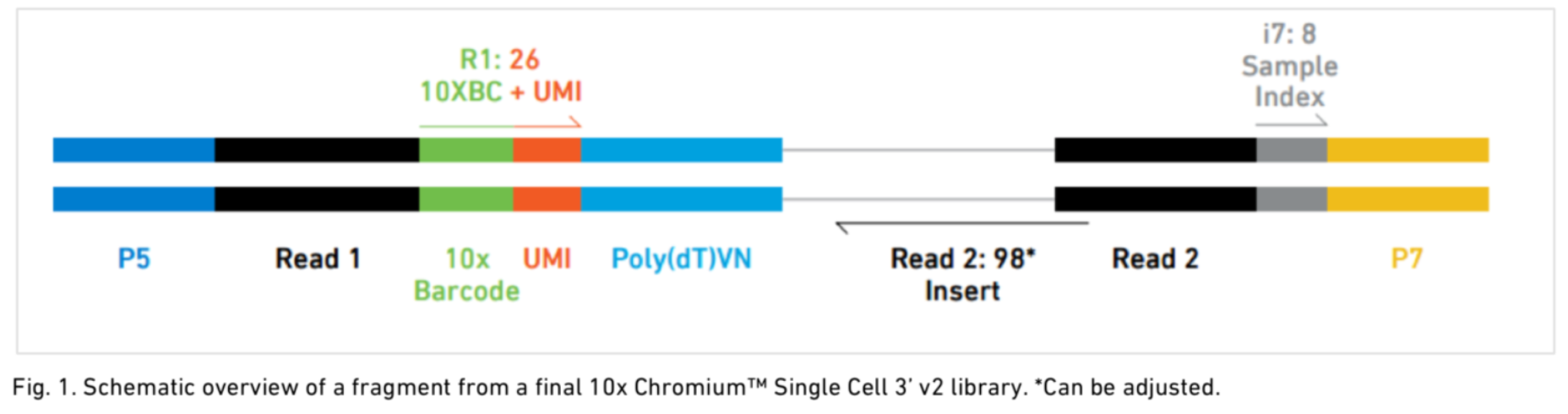
4.2 10X Barcode(Cell barcode)的作用?
10X Barcode(Cell barcode)是10X数据特有的,用来区分GEMs,可对细胞做了一个标记。
4.3 UMI的作用?
在scRNA测序中需要进行PCR扩增, 一些转录本会被扩增多次,超过了其真实的表达量。当起始文库DNA量很小时,在进行多次PCR扩增中,引入的误差会随着扩增次数的增加而增加。
UMI - Unique Molecular Identifier, 由4-10个随机核苷酸组成,在mRNA反转录后,进入到文库中,每一个mRNA随机连上一个UMI,根据PCR结果可以计数不同的UMI,最终统计mRNA的数量(重点)。
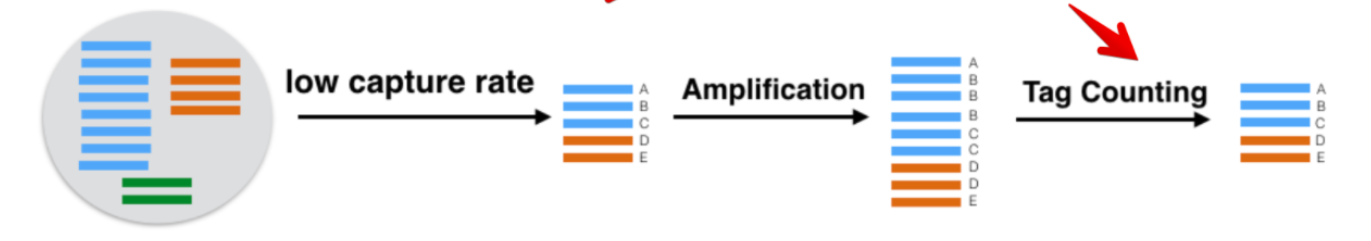
对UMI的要求:
- 不能是均聚物 ,如AAAAAAAAAA
- 不能有N碱基
- 3.不能包含碱基质量低于10的碱基
4.4 简而言之
Library Barcode (Sample Index) : 使用样本index序列进行多样本拆分
10x Barcode(Cell Barcode ): 用来区分细胞reads的来源
Unique Molecular Index (UMI) : 用来校正PCR扩增引起mRNA数量统计的偏差
Sequencing Reads : 用来识别基因的reads
5. fastq文件重命名
参考以下命名要求,对SRA拆分获得的样本fastq.gz文件进行重命名。

# 按以上规范,执行bash批量重命名
cat SRR_Acc_List.txt| whilre read srr;do \
(mv ${srr}_1*.gz ${srr}_S1_L001_I1_001.fastq.gz; \
mv ${srr}_2*.gz ${srr}_S1_L001_R1_001.fastq.gz; \
mv ${srr}_3*.gz ${srr}_S1_L001_R2_001.fastq.gz);done






 苏公网安备32011502012024号
苏公网安备32011502012024号