单细胞RNA测序入门可以查看以下文章:
单细胞RNA测序(scRNA-seq)工作流程入门
1. 单细胞的分离
如何获得单细胞,从而进行下一步的测序过程?具体有以下几种方法:
一、流式细胞仪(FACS)方法
常用的方法之一是使用流式细胞仪(FACS)方法进行分离。但是 FACS 也有缺点,首先需要较大的起始细胞量,同时可能导致有些孔中有两个,有些孔中没有细胞。
二、显微操作分离
显微操作提供了获得少量细胞的替代方法,我们可以通过直接特定位置摘取单个细胞进行测序,但是这种方法依赖于人工操作,无法实现高通量进行。
三、微流控技术
近些年微流控技术在单细胞分选上展现良好的应用,微流控技术可以对整个过程进行监控和处理,其缺点主要在于固定的微流控芯片等。通过多通道微流控自动处理技术进行处理,可以允许自动分离和处理细胞,同时控制细胞的数量和实验的误差。
2. 全基因组扩增
对于一个细胞(哺乳动物细胞)而言,通常细胞DNA不到10pg,需要先对全基因组进行扩增才能开展后续的实验。全基因组扩增技术(WGA)主要是通过 PCR、多重替换扩增技术(MDA)或两者的组合来实现。
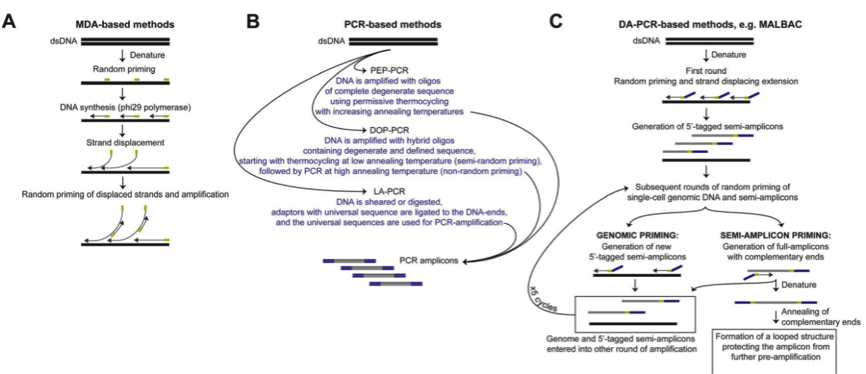
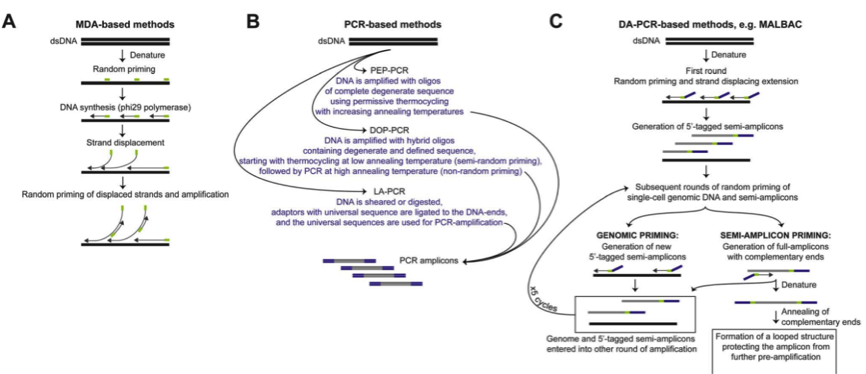
一、PCR扩增策略
PCR 策略主要是通过使用随机引物进行扩增,然后引入通用接头(adaptor)。随机引物 PCR 方法相对于MDA,有更高水平的一致扩增, 但是覆盖度较低。
二、多重替换扩增技术(MDA)策略
MDA 方法则利用具有链置换活性的 DNA 聚合酶(一种滚环复制酶),进行恒温扩增, 可以处理长达 10 Kb 的序列,一旦接触到前面结合的序列后,聚合酶可以释放前者 DNA,从而释放用于进一步的扩增。MDA 方法由于 DNA 聚合酶的高保真性和高处理特性,可以实现较好的覆盖度也有利于 SNP 等精细实验,然而由于 GC 含量等影响,MDA 可能导致不一致扩增,Gcbias 比较大,因此 MDA 用于 CNV 检测等的假阳性概率比较大。
三、MALBAC 策略
华人科学家谢晓亮等人基于前两种方法提出综合两种方法的 MALBAC 方法,实现了高覆盖度的一致性扩增方法。
通过增加互补末端引物使用滚环复制酶产生可以末端配对的 loop,被扩增出来的 DNA,由于末端配对,因此无法作为接下来的模板,从而可以实现近乎线性的扩增,经过五轮线性扩增后,然后通过 PCR 进行指数级扩增,相对于 MDA 方法 31%-61% 的丢失率,MALBAC 方法只有 1% 的丢失率,同时对于 SNP 和 CNV 的检测的假阳性更低。
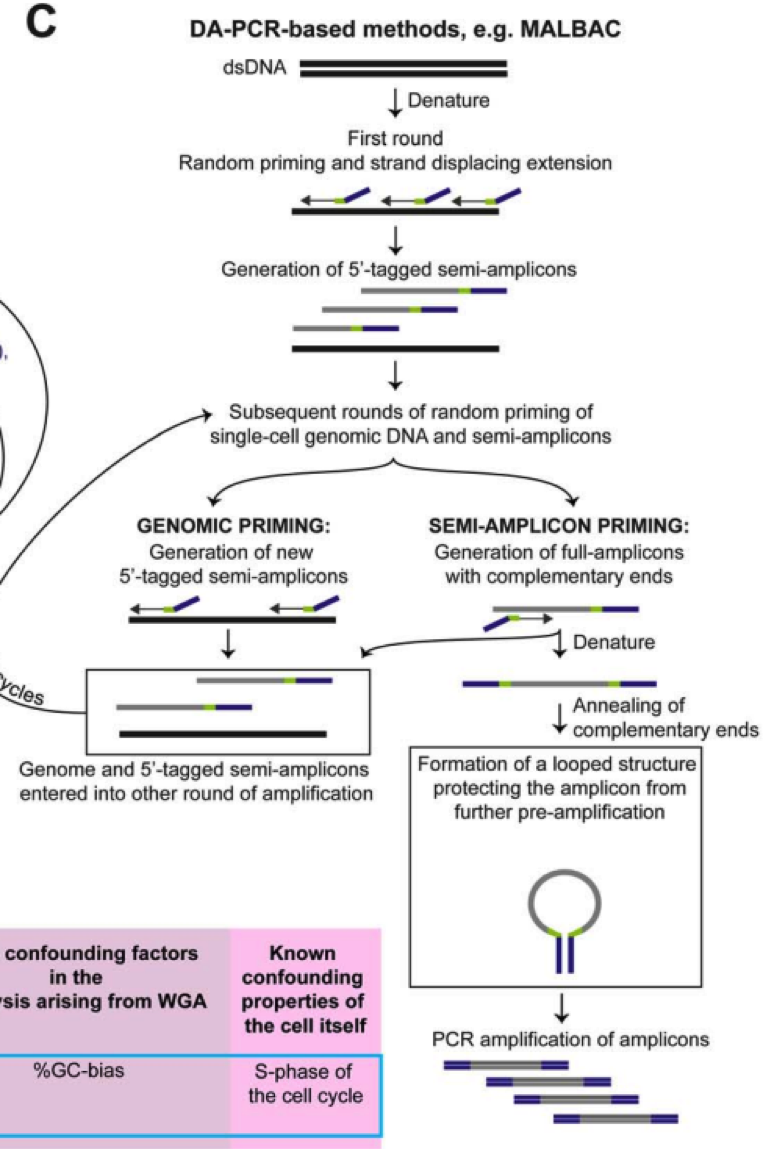
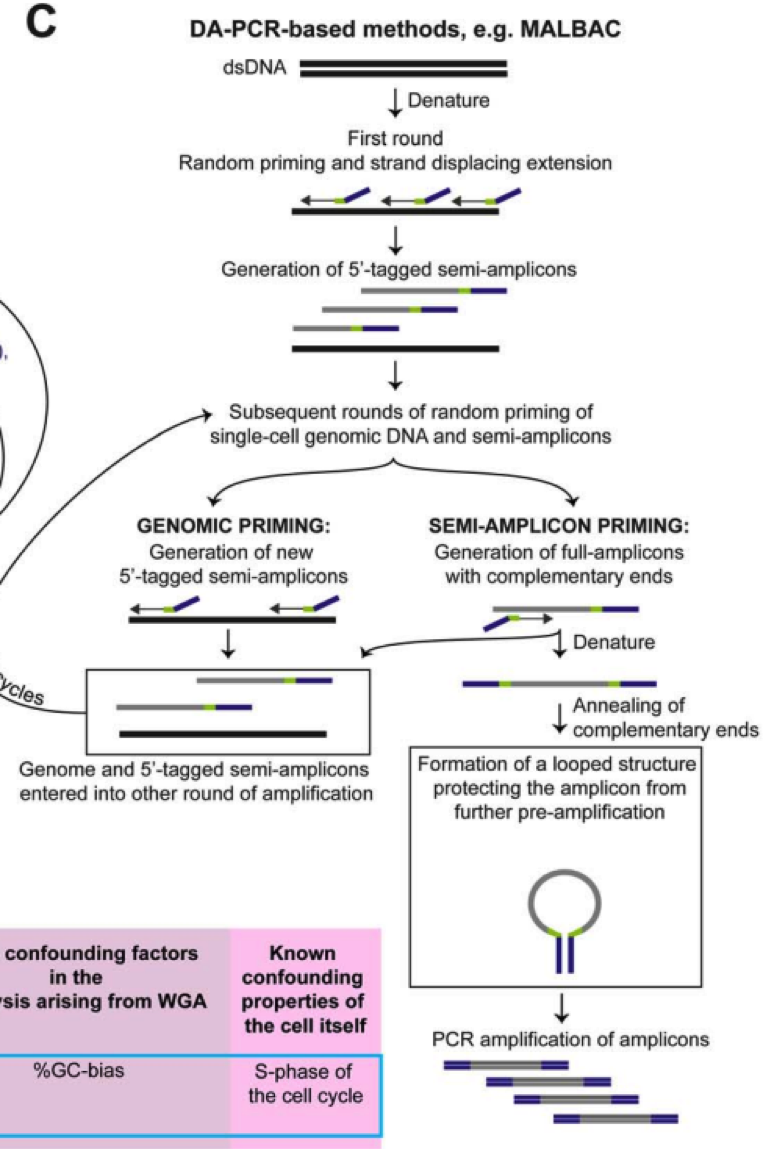
3. 单细胞DNA-seq基本工作流
单细胞基因组测序后,需要剔除低质量的reads,比对到参考基因组,校正由GC偏好引起的reads数的误差,后续可以使用GATK工具进行Variant calling。
不同单细胞 DNA 测序的方法特征对比
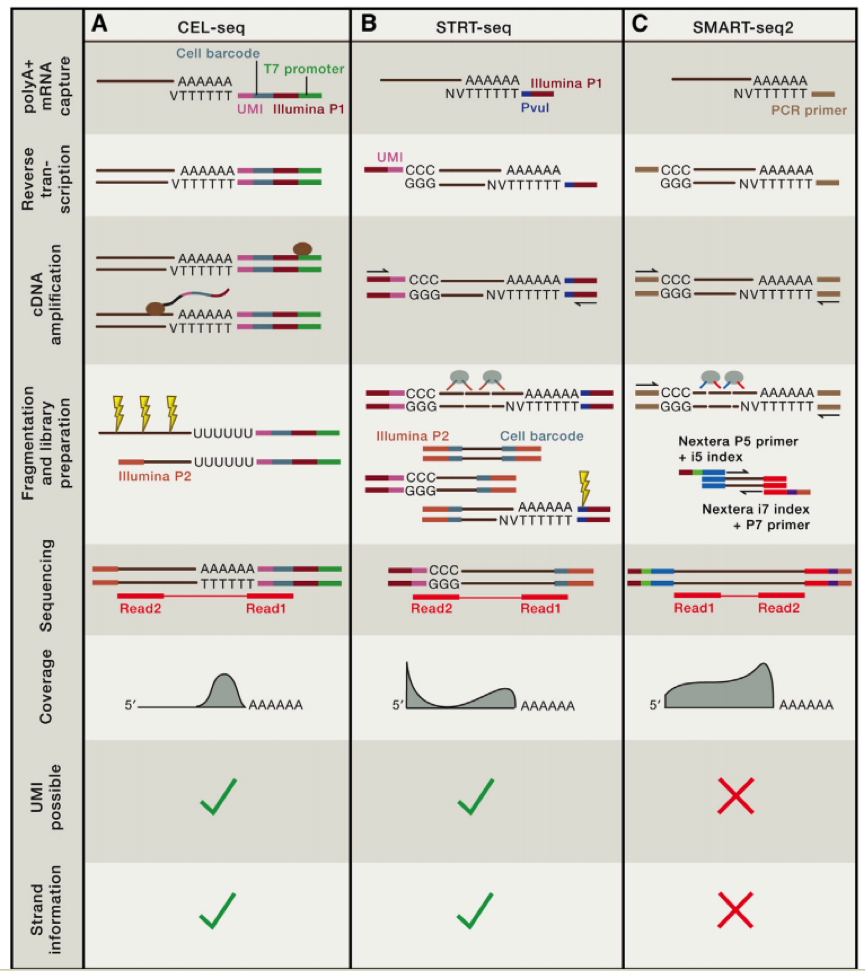
4. 单细胞RNA-seq基本工作流
单细胞 RNA-seq 的主要障碍是如何从单个细胞中获取足够的 RNA 用于下一步的测序,现有多种的方法扩增得到低于 pg 级的 mRNA。扩增mRNA的主要障碍是扩增的偏好(bias)问题,其会对来自不同基因的 mRNA 之间的差异比较产生干扰。
一、单细胞RNA-seq扩增技术的发展
利用单细胞进行 RNA-seq 是 Surani 实验室在 2009 年开发出来的,扩增的方法主要是使用将 RNA pull down 下来,然后使用 polyT 引物反转录带 ployA 的 RNA,同时引物上含有特定的 anchor 序列。
研究人员借鉴了基于 microarray 用于 single cell sequence 的技术后,修改了一些实验条件(比如延伸时间等),从而获得全长的 cDNA,进一步通过 SOLID 测序,我们可以看到具有非常好的稳定性。
2011 年科学家们使用了新的 cell barcode 方法,从而可以探究多个复杂细胞的特性,称为 STRT 技术,但是 STRT 技术对于 5’end 具有偏好性,基于转座子酶,人们开发了升级版的 STRT2 技术。
为了减少由于扩增引起的误差,人们在一些单细胞测序的步骤中增加了 UMI(unique molecular identifiers),UMIs 是由 4-10 个随机核苷酸组成的序列组成,在 mRNA 反转录后,加入到文库中,每一个 mRNA随机连上一个 UMI,因此可以计数不同的 UMI达到计数 mRNA 数量的目的。
二、单细胞RNA-seq扩增技术的选择
如果我们想了解不同细胞间的异质性,那么需要减少实验的 bias,推荐使用 UMI 相应的技术,如果我们想了解 RNA 全长,包括 剪切位点 等信息,那么我们就需要选择包含全部转录长度的技术。
三、UMI计数
如果我们使用 UMI 计数方法( UMI 计数,是指在建库过程中,通过在引物上,增加随机序列,则对于同一种 mRNA 连上同样的 UMI 概率几乎为 0,则可以忽略由于 PCR 造成的误差,对于一种 mRNA,测到的 UMI 数量可以近似看成 mRNA 的表达个数),推荐至少每一个转录本至少覆盖 3-4 倍,确保一些低表达的 mRNA 不会因为 噪声而被忽略,一般而言,illumina 的一条 lane 中最终可以被使用的数据大概占 50%,另一些 reads 因为实验技术所限,为一些 接头adaptor 等无效序列。
假设想获得1000个转录本信息,以nextseq一个lane可获得200M条reads,需要进行检测细胞数量则= 200,000,000 / 1000 / 4 / 2 = 250个细胞。
5. 单细胞RNA-seq的数据挖掘
单细胞 RNA-seq 为我们了解组织的组成等提供非常好的工具,而单细胞测序的效果也取决于我们到底测多少细胞,一般而言几百个细胞的单细胞测序不仅仅捕获常见的细胞成分,也会捕获稀少类型的细胞。由于 RNA 降解,细胞的凋亡和实验误差等原因,一些数据将会被排除,不纳入数据分析环节,因此初始选择的细胞个数往往会大于我们目标测序的细胞个数。
对于不同转录本,没有要求的话,推荐大家使用多转录本合并的序列。因为大部分单细胞测序主要是富集 3‘UTR 部分,那么如果使用 cufflink,就会造成很大的问题,比对完后,需要确定每个转录本的表达量,前面我们说到UMI用于 RNA 的计数,在计数前,还需要对不同的细胞进行滤过。
由于存在 RNA 降解等多种问题,需要考虑到去除一些低转录本等情况,同时还需要考虑去除低扩增效率的细胞。需要做的便是归一化,进而可以在不同细胞之间的进行横向比对。将所有的 cell 归一化到 reads 总数是中位数的细胞的 reads 总数中,或者将所有细胞归结成同样的 reads,被称为 down-sampling。
在获得不同细胞的不同基因的表达矩阵后,便可进行细胞分类注释及下游分析。*






 苏公网安备32011502012024号
苏公网安备32011502012024号