
「一边学习,一边总结,一边分享!」
一、 写在前面
2023年4月中旬自己开始做基因家族的分析,对于这块自己没有接触过,因此也是一个挑战,没事!!!(安慰自己),对于基因家族的分析网上的教程很多,跟着步骤走就可以。在这部分,我自己主要是做生信这块,实验验证是师姐在做,所以论文结构自己不用操心。此外,可视化的工具很多,也很方便,不需要自己特意去学。
此外,本次分析80%的内容都是基于TBtools。确实牛X!!自己开始接触TBtools是在2019年吧,也是通过一个师兄的推荐才知道的。2019年CJ还没将TBtools发表在MP上,那时还是预印版本吧。但是,引用已经有了很多,了不起哦。后面TBtools一直在开发新的的小“软件” or“程序”,将生信分析的门槛一降再降。点赞点赞!!! --Du
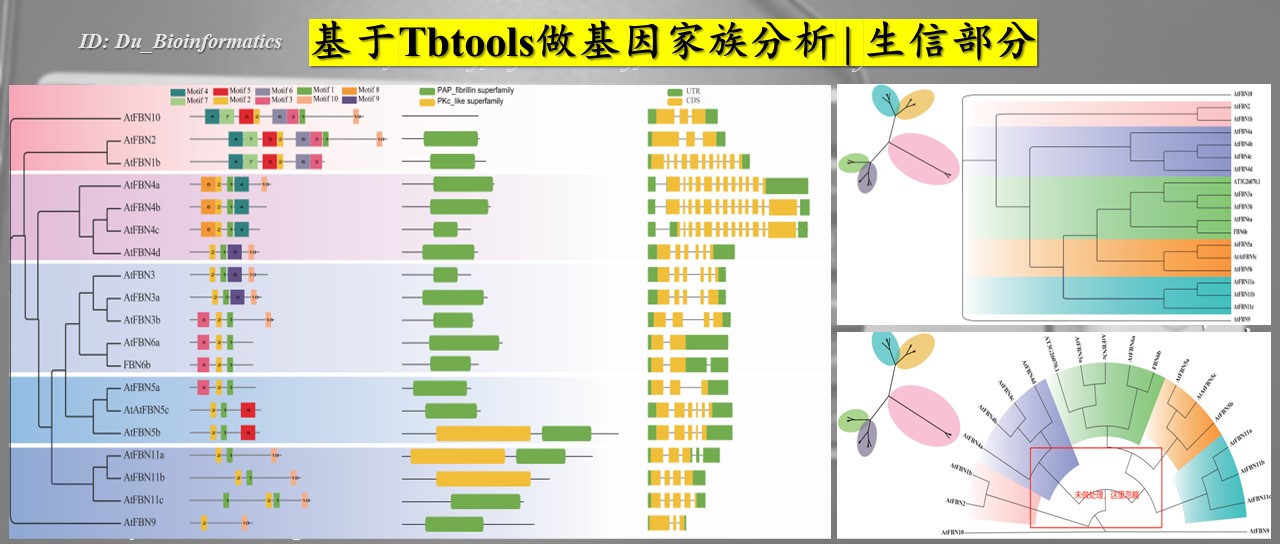
注意:此教程有些话语可能会带有自己的方言,读不通时也不要在意!![泪目!]
一,在Pfam数据中获得基因家族
我们这里预测作物中某一个基因家族的基因,目前在此作物中未报道。因此,使用Pfam数据库中一致的基因进行同源搜索(其实,你也可以使用已知作物中的基因进行同源搜索,获得结果基本一致)。那么我们就根据文章中和报道的Pfam数据库中的基因作为基序,进行同源搜索。
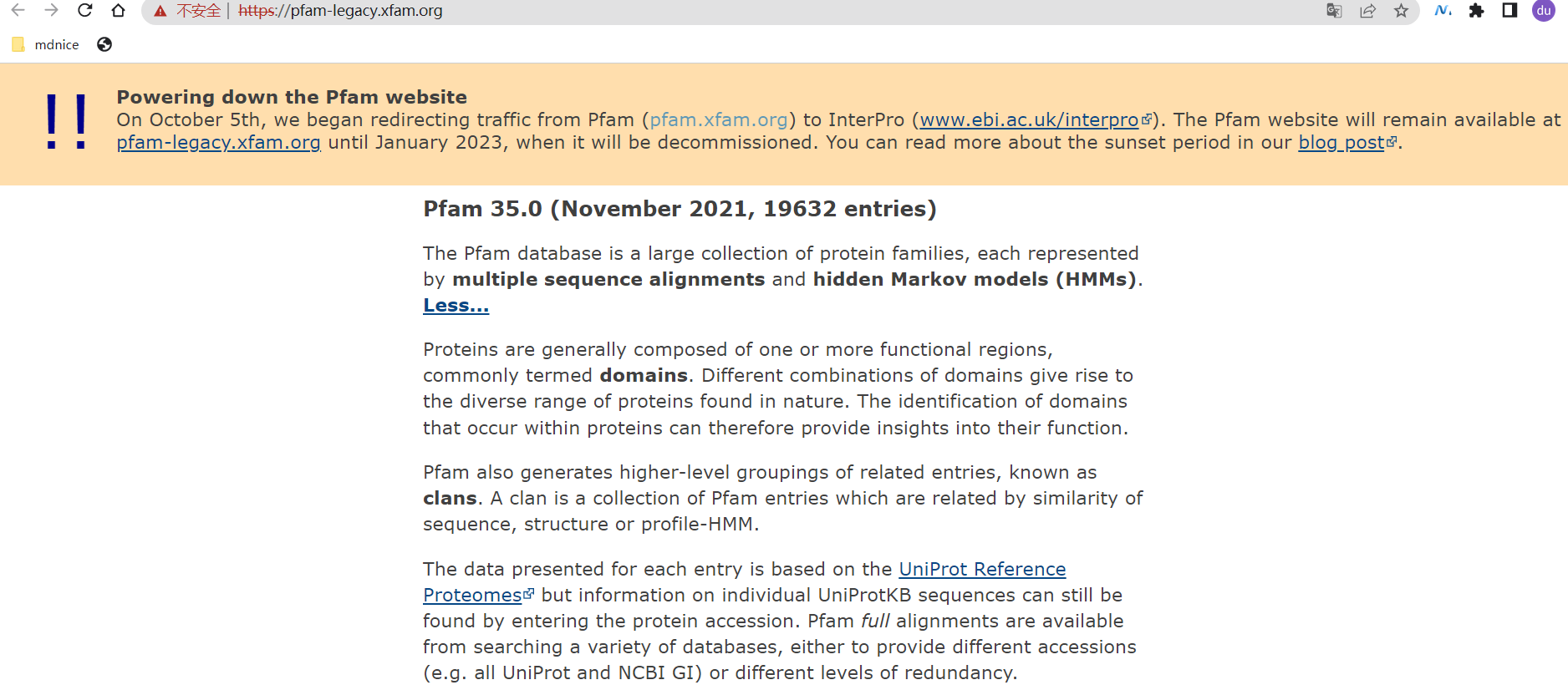
- 在Pfam数据库中下载FBNs基因家族(Pfam 04755),Pfam网址:https://pfam-legacy.xfam.org/

- 打开网址:http://www.ebi.ac.uk/interpro/entry/pfam/?search=0477#table
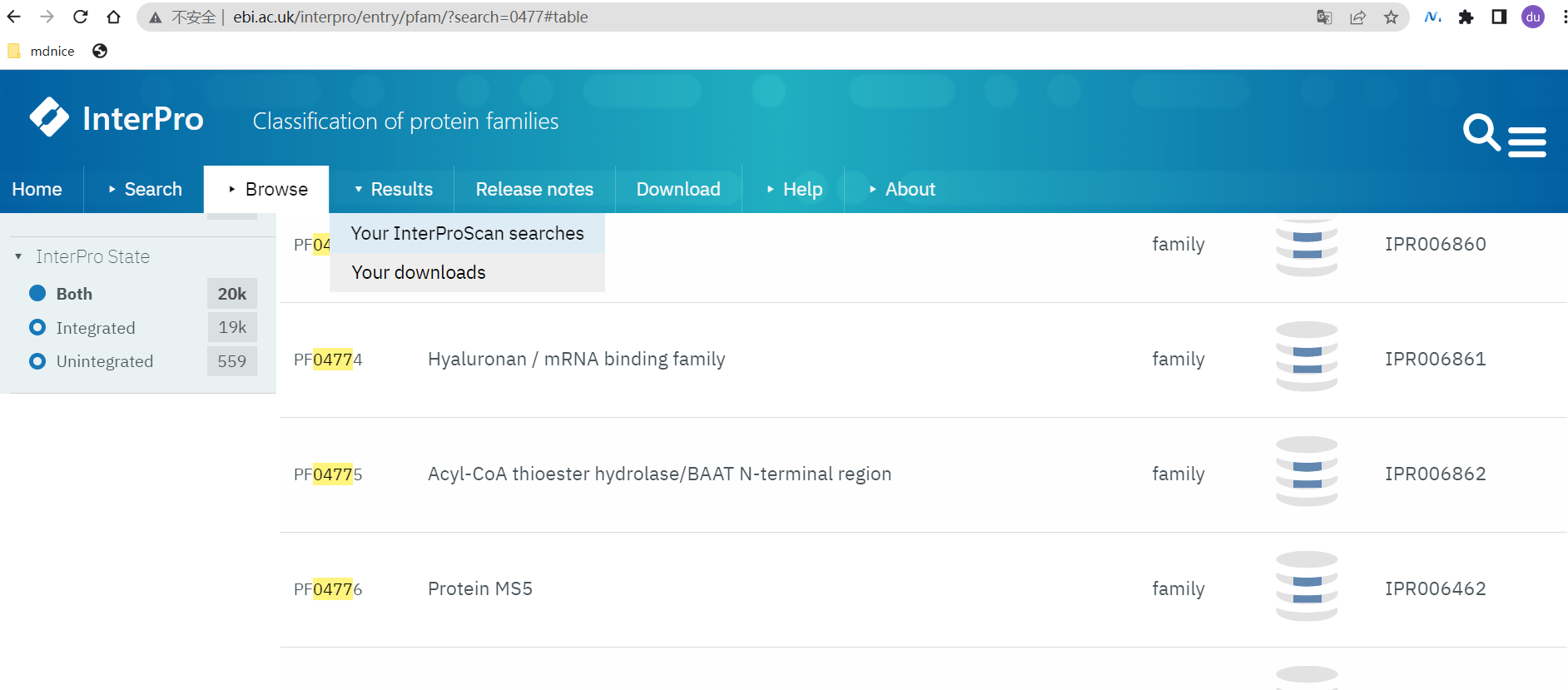
- 点击进入PF04775,下载所有的Proteins序列

以上只是其中的一种方法,但为获得FBN基因家族的蛋白序列。下面使用Pfam数据中搜索
- 打开网页。https://pfam-legacy.xfam.org/
- 搜索
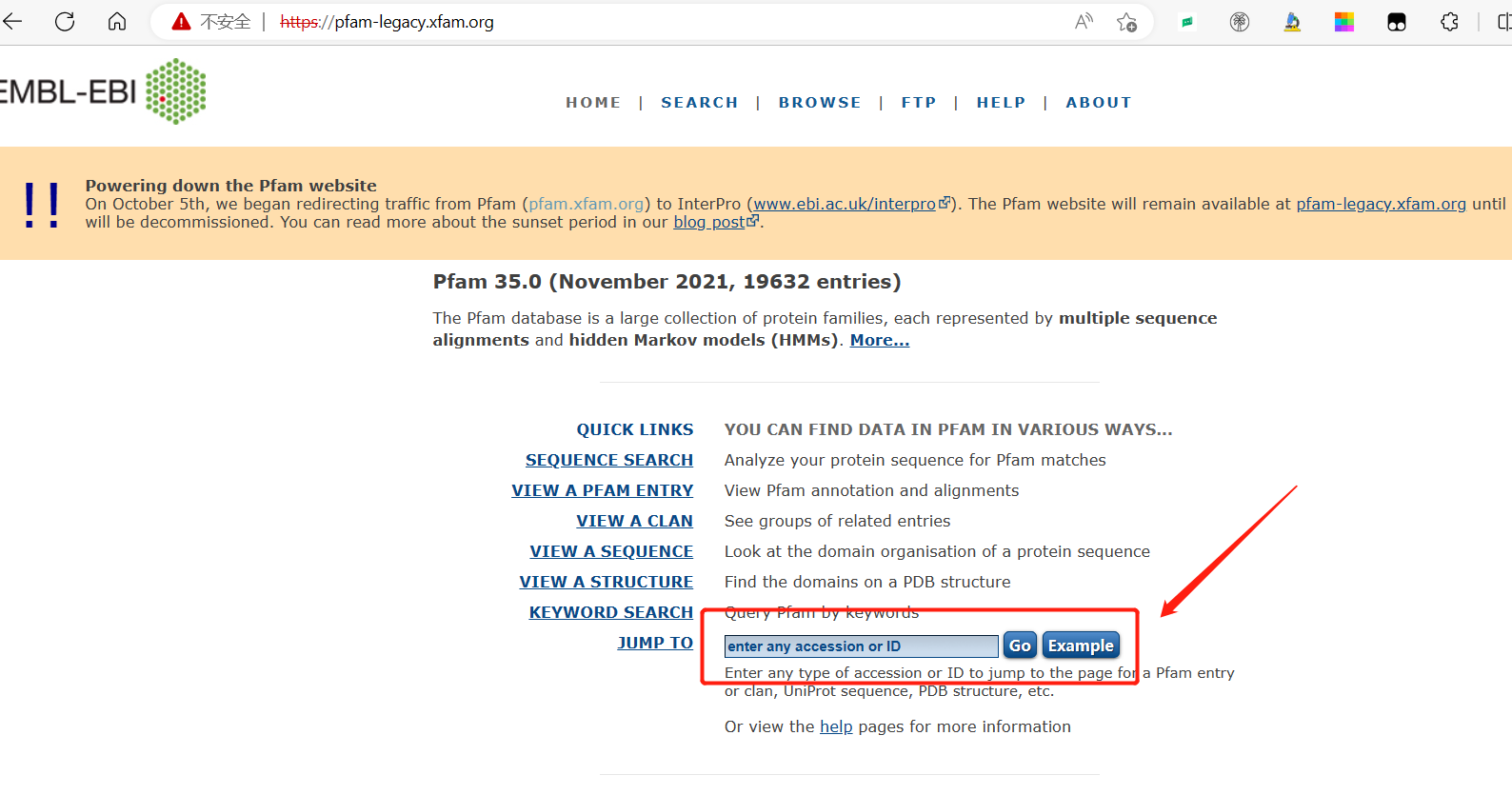
- 进入
- 搜索后获得PAP_fibrillin
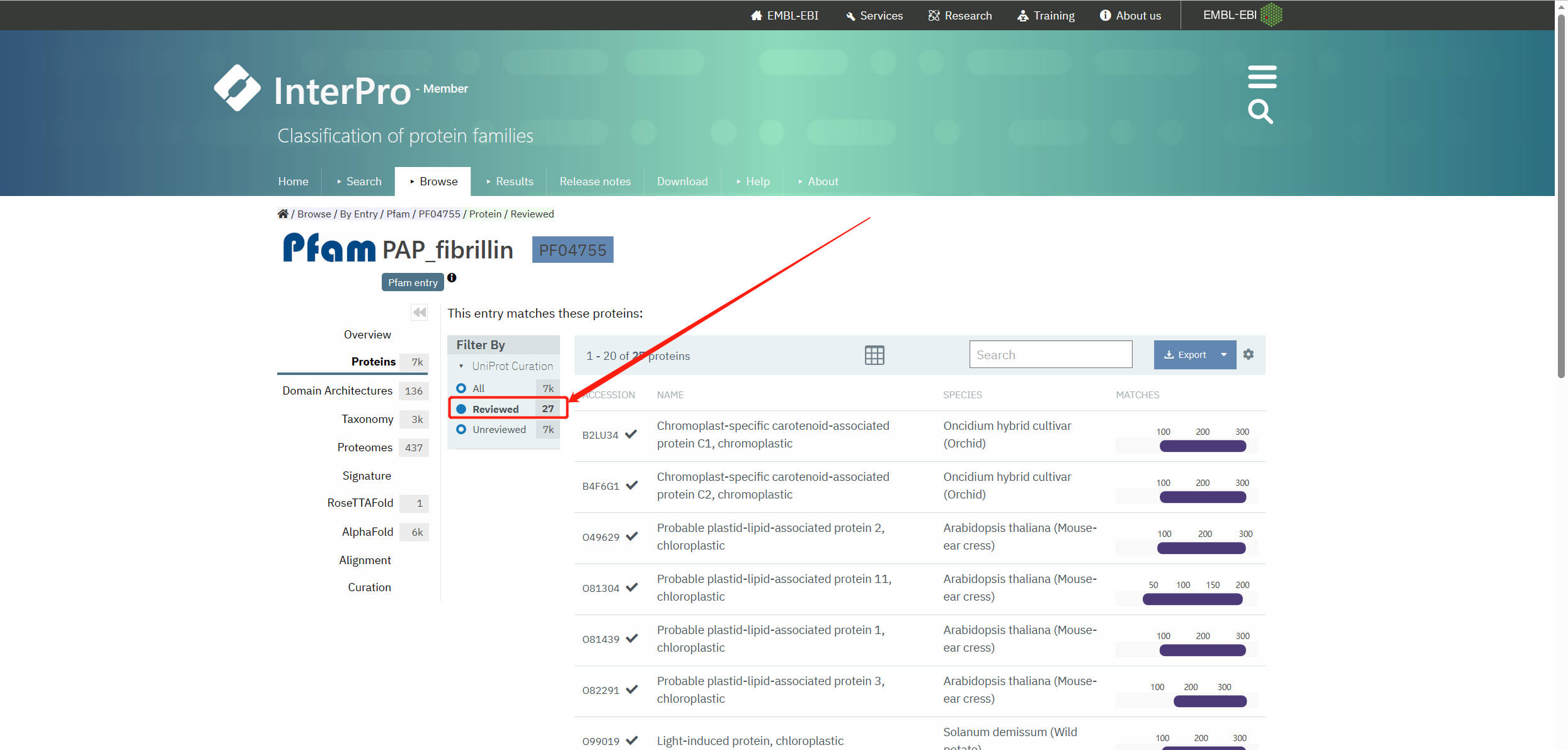 下载Reviewed的PF04755序列
下载Reviewed的PF04755序列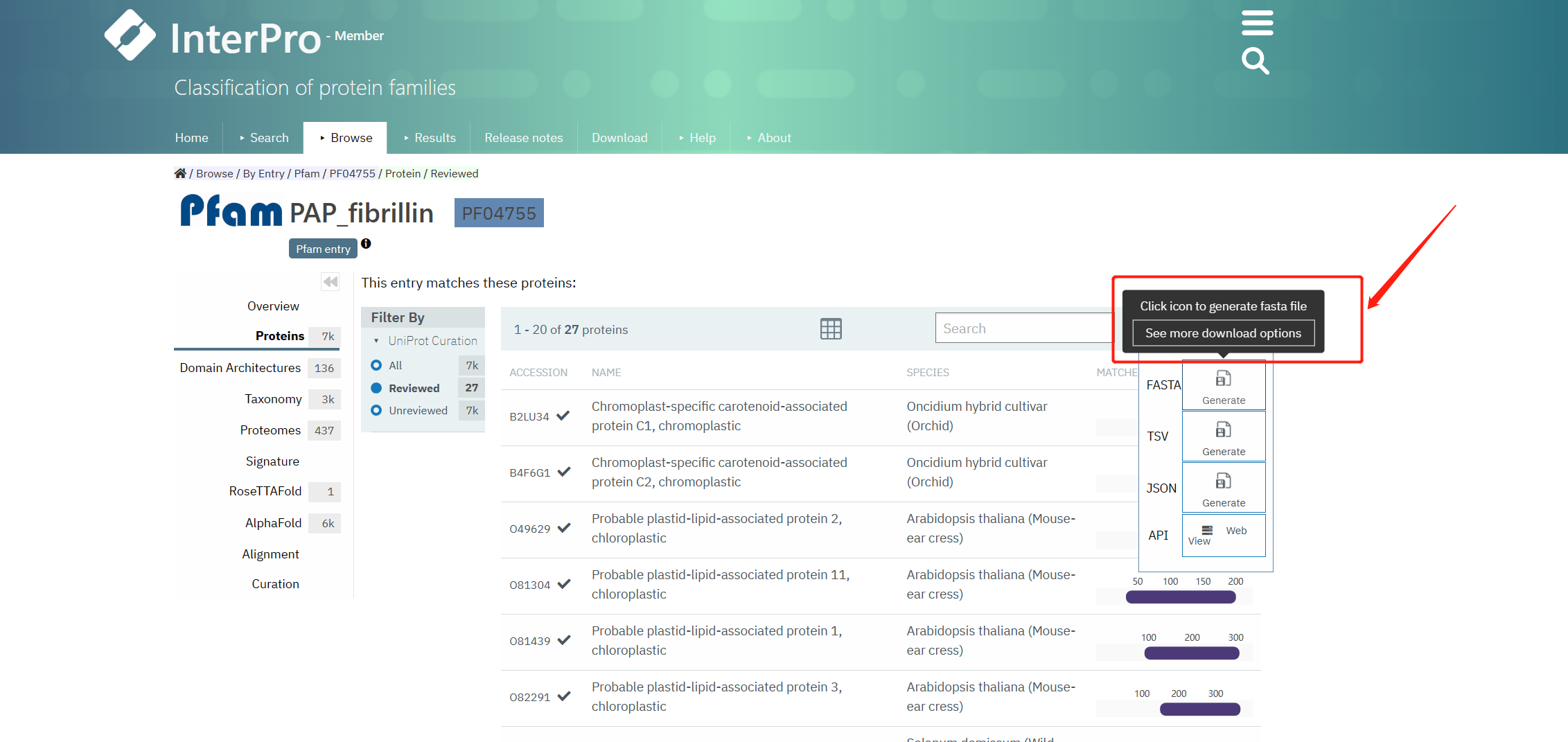
二、同源序列检索预测
对于同源基因的搜索,很多基因家族的文章都使用HMMER进行检索,也有一些文章是使用BLAST。你任选其中一个即可,都能获得你想要的结果同源基因。在做分析的时候,我将使用Hmmer寻找同源基因的文章分享在公众号中,在评论区有一个大佬对HMMER和BLAST之间的差异给出回答。
这两个方法原理上区别,balstp是基于序列同源性进行打分的,有打分矩阵,hmm是基于隐马尔可夫模型,对序列结构域进行比对。**来自“泼皮混混”的评论。**
2.1 HMMER同源结构域搜索
2.1.1 Hmmer的安装
安装,主要是使用源码安装或是是使用conda进行安装即可。
- conda安装
conda install -y hmmer
- 源码安装: 官网**:http://www.hmmer.org/**任意下载一个版本即可,安装步骤不再做说明。
2.1.2 使用hmmbuild构建.hmm文件
在有些数据库中是有.hmm文件,只需要下载即可。但是,这仅仅只限于有些大数据库。对于我们自己使用,不可能全部都有,这就需要我们自己构建,**很多教程到这步就是让你收费了.......**。
在本教程,讲述其中一种方法吧,希望对大家有所帮助。
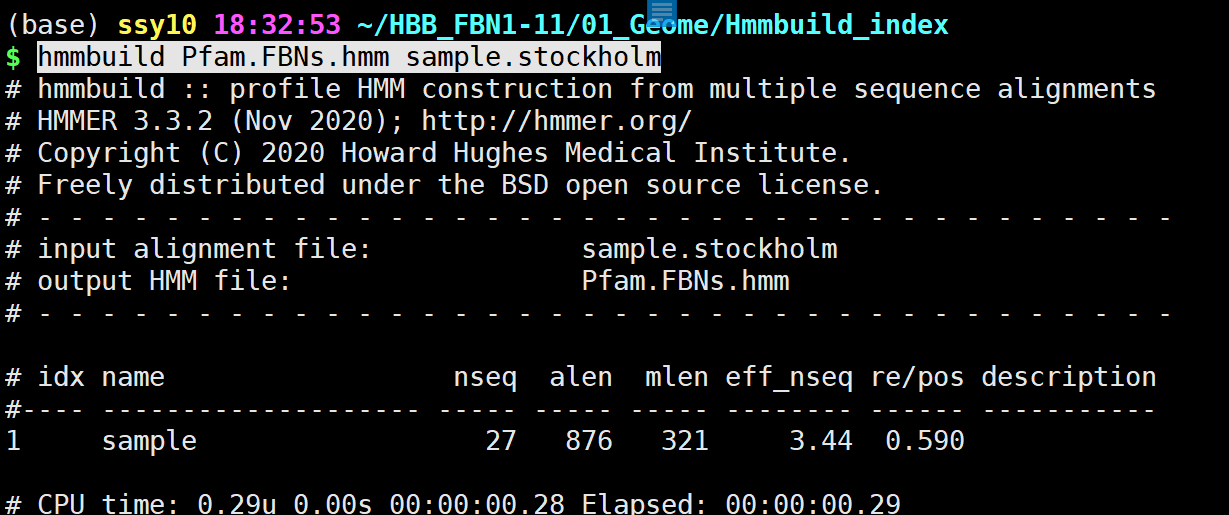
hmmbuild构建时,需要使用.sto文件进行构建。因此,我们必须获得.sto文件。
- 使用mafft软件进行间序列进行对齐
mafft --auto --clustalout ../Pfam_PF04755_reviewed.fasta > Hmmbuild_index/Pfam.FBNs.align.clustal
 转换: http://sequenceconversion.bugaco.com/converter/biology/sequences/fasta_to_phylip.php
转换: http://sequenceconversion.bugaco.com/converter/biology/sequences/fasta_to_phylip.php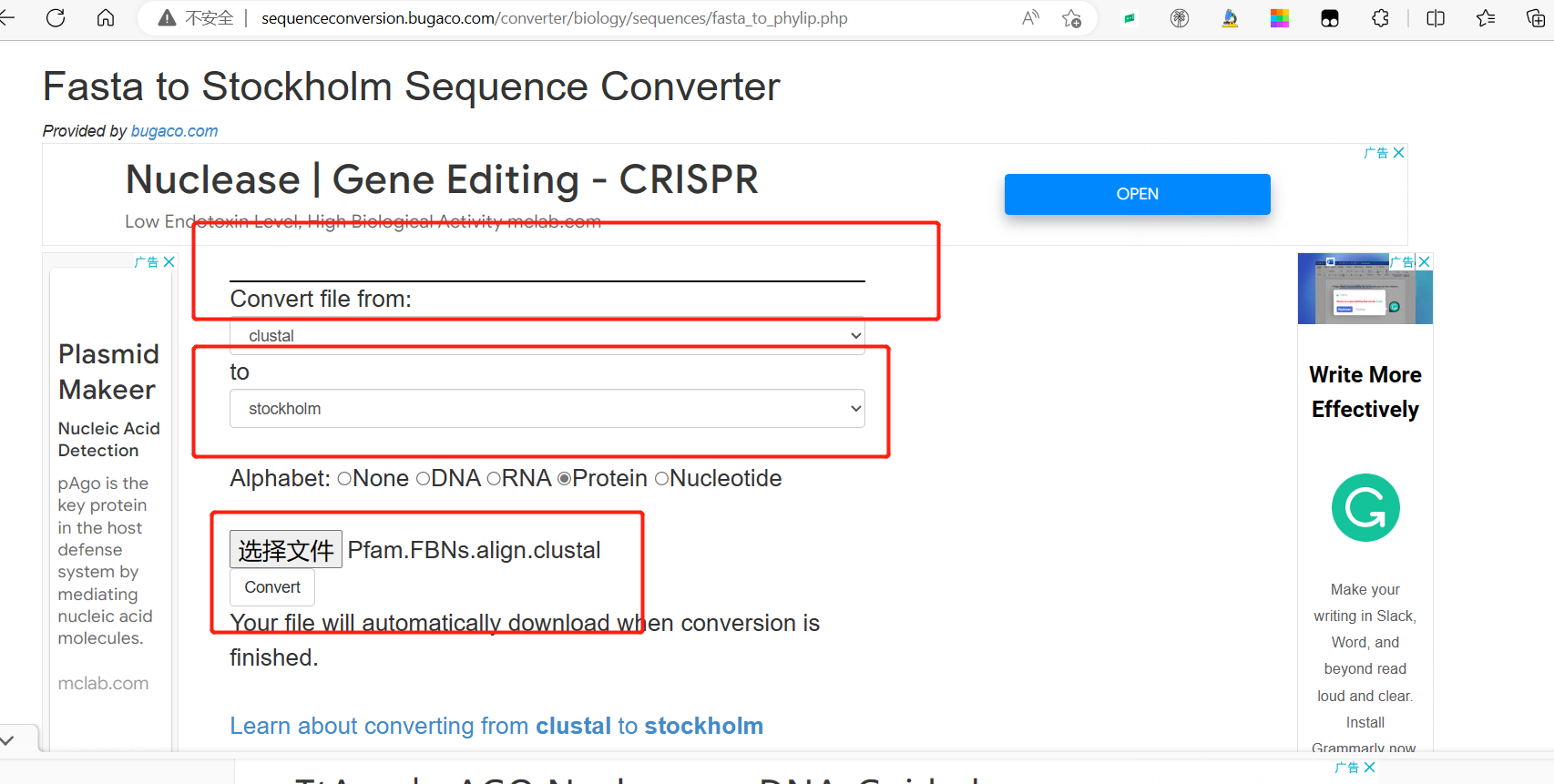
hmmbuild Pfam.FBNs.hmm sample.stockholm
hmmsearch Hmmbuild_index/Pfam.FBNs.hmm Potato/DM_1-3_516_R44_potato.v6.1.working_models.pep.fa > ../02_Result/Potao.hmmer.out.txt

- 筛选出最佳的结果,E-value值小于1e-5,Score值大于“> 90”
- 对于筛选结果,可以直接使用Hmmsearch获得结果;也可以如上所示根据自己需求进行筛选,自己做的话,如果搜索的目的基因太多,而自己不需要这么多的同源基因,自己会进行手动过滤一些同源性较弱的基因。
cat Potato.hmmer.out.txt |grep -v "#" | awk '{if($4 < 1e-5 && $5 > 90) print $9}' | sort | uniq | grep -v "+" > Potato.hmmer.best.out.txt
2.2 提取目的基因序列
日志:通过Hmmsearch获得同源基因的ID,那么后面对目的同源基因进行进化树、结构域、motif等的分析,这些分析都需使用目的同源基因的序列。
如何获得同源基因序列??
- 使用脚本获得
- 使用ggffead获得,需要获得同源基因的.gtf文件等信息。
- 生信工具获得、如TBtools等。
对于这步、我们就多做讲解,使用自己拿手的方式获得即可。
问:**后面的分析使用核酸序列 or蛋白序列呢??**
答:**都可以。**
FBN 家族的分析日志。使用Pfam、拟南芥(11)和水稻的FBN家族基因同源搜索马铃薯中的FBN同源基因
## 水稻中的FBN家族基因
cat all.pep | grep ">" | grep fibrillin |awk -F "|" '{print $1}' | awk -F " " '{print $1}' | sed 's/>//g' > O_sativa.FBN.id.txt
##拟南芥中FBN家族基因
可以在拟南芥网址中的同源搜索,也可以在拟南芥蛋白数据中搜索
cat Araport11_pep_20220914 | grep FBN | awk -F "|" '{print $1}' | sed 's/>//g' > Araport11_FBN.id
2.3 使用TBtools提取目的基因
说实话,TBtools确实是个很牛的生信工具,基本可以让你不写代码获得你想要的东西。以及,各种类型的小脚本软件都一直在开发。赞赞!!
2.3.1 TBtools软件的下载
- 网址:https://github.com/CJ-Chen/TBtools
- 安装。
- 动手运行
2.3.2 提取序列
- 准备作物所有的蛋白序列文件(or基因文件)
- 目的基因的ID
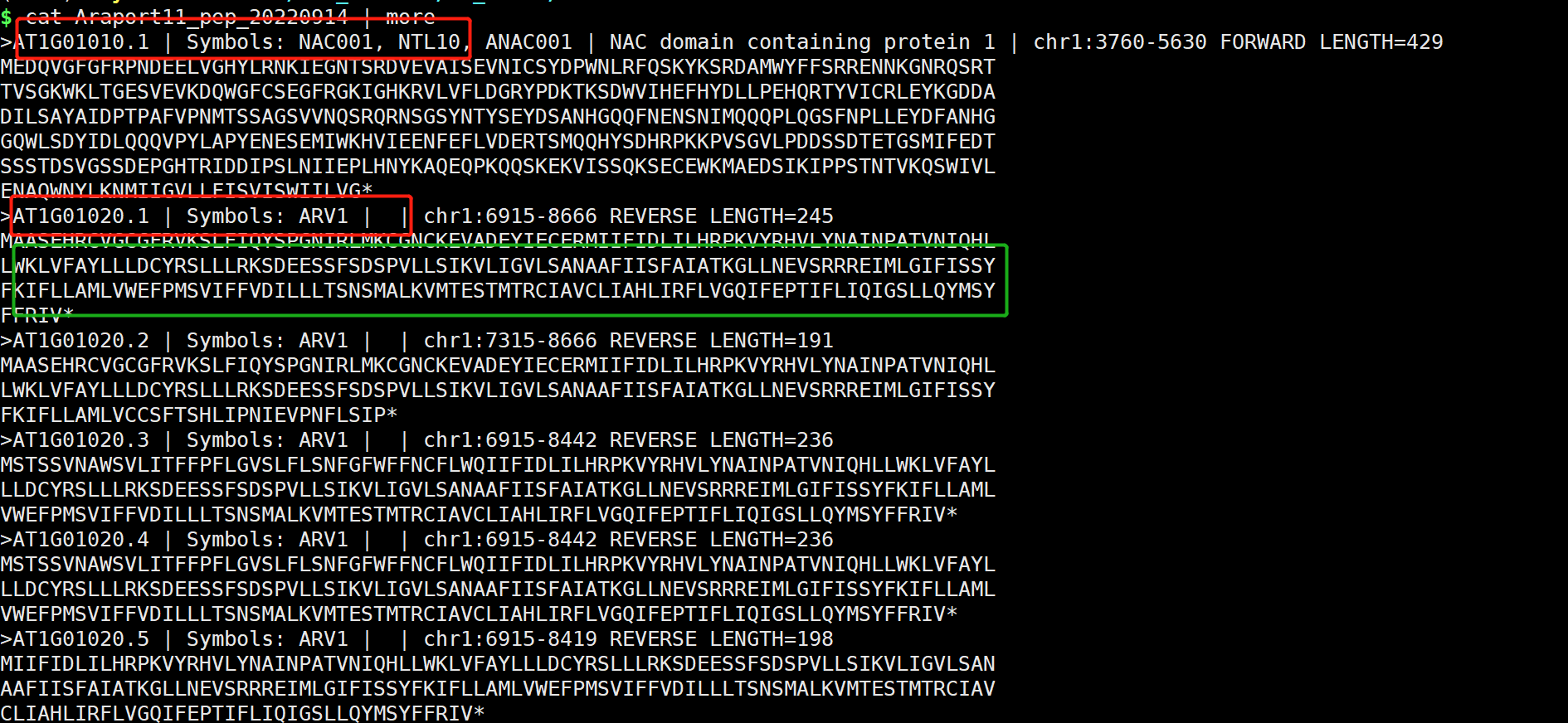
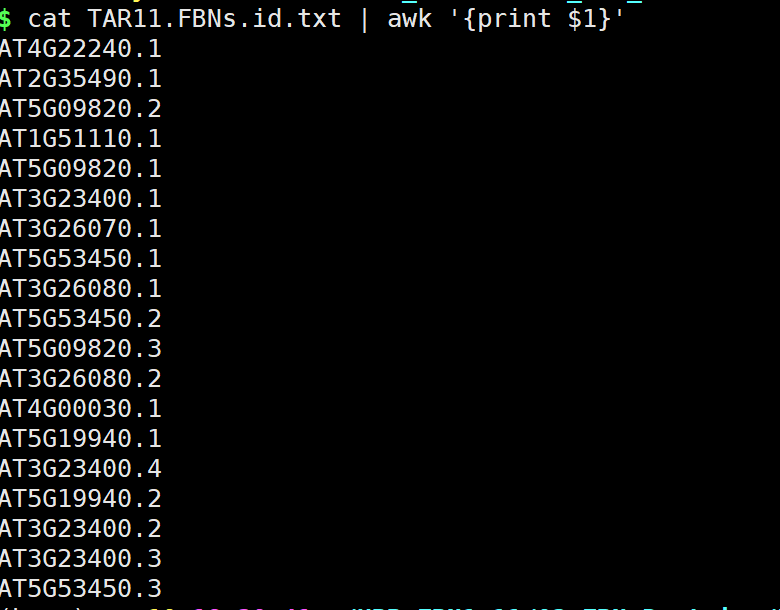
- 打开TBtools,Fasta Extract or Filter (Qyick)

- 获得结果
2.4 目的同源基因motif分析
2.4.1 使用MEME进行motif预测
- 网址:https://meme-suite.org/meme/tools/meme

- 上传相关的fa文件,以及修改相关的参数,进行提交
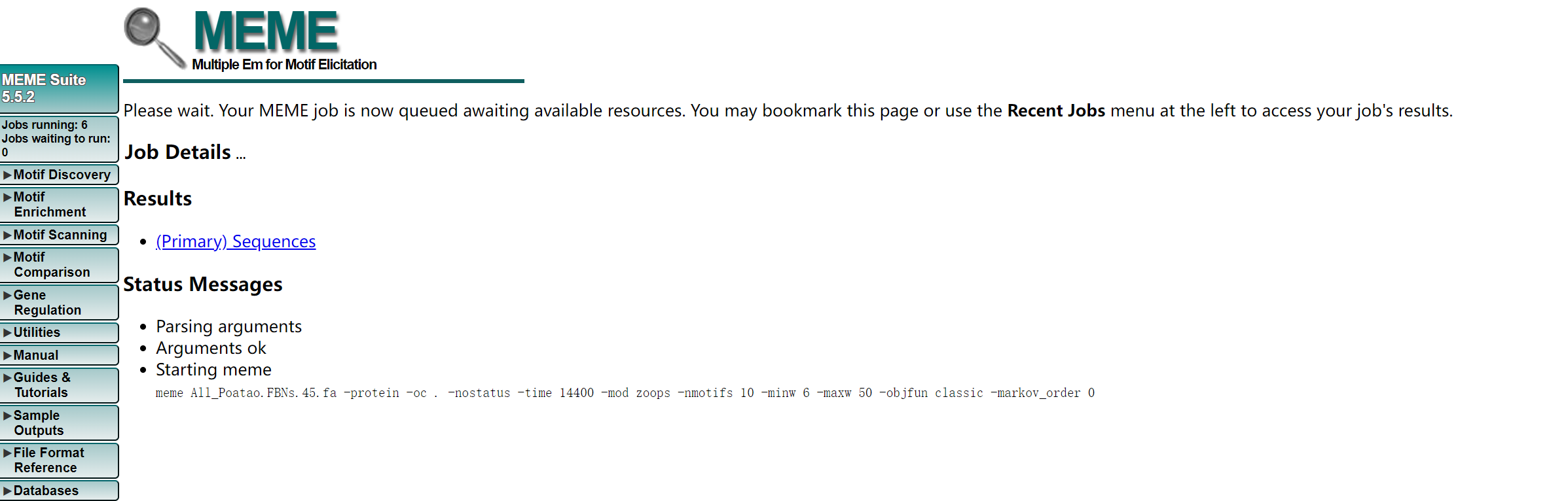
- 输出结果
 输出结果很快,有以下几个结果文件。
输出结果很快,有以下几个结果文件。
2.4.2 motif可视化
对于motifi分析可以参考一下文章:
- TBtools | 多图合一至强版教程!进化树 + Motifs + 结构域 + 启动子 + 基因结构 + ....,TBtools开发者本人的教程
- TBtools | 基因家族分析 (进化树、Motifs、结构域)
- 或是本篇教程
MEME网址结果可以给我们的seqlogo信息和motif信息。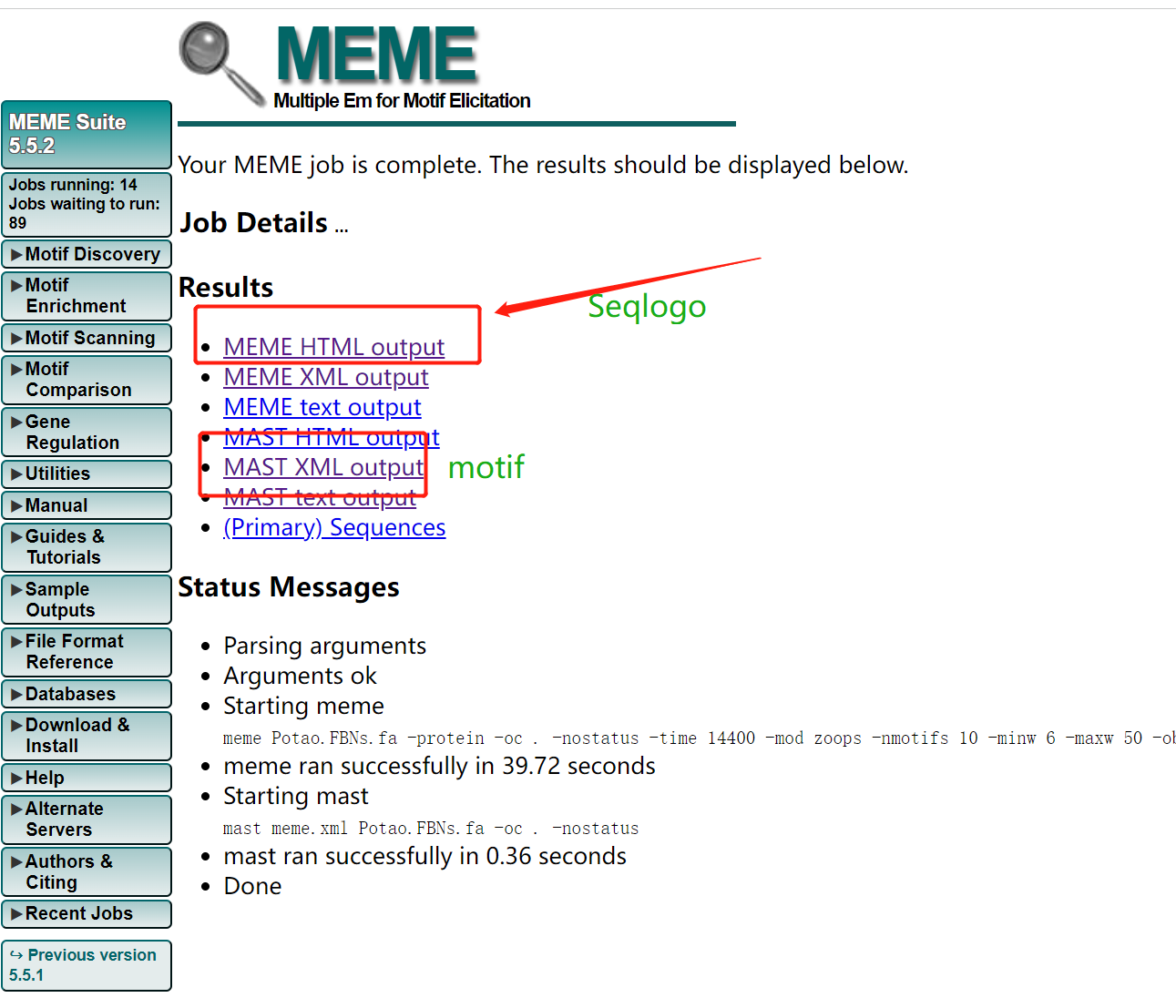
- Seqlogo
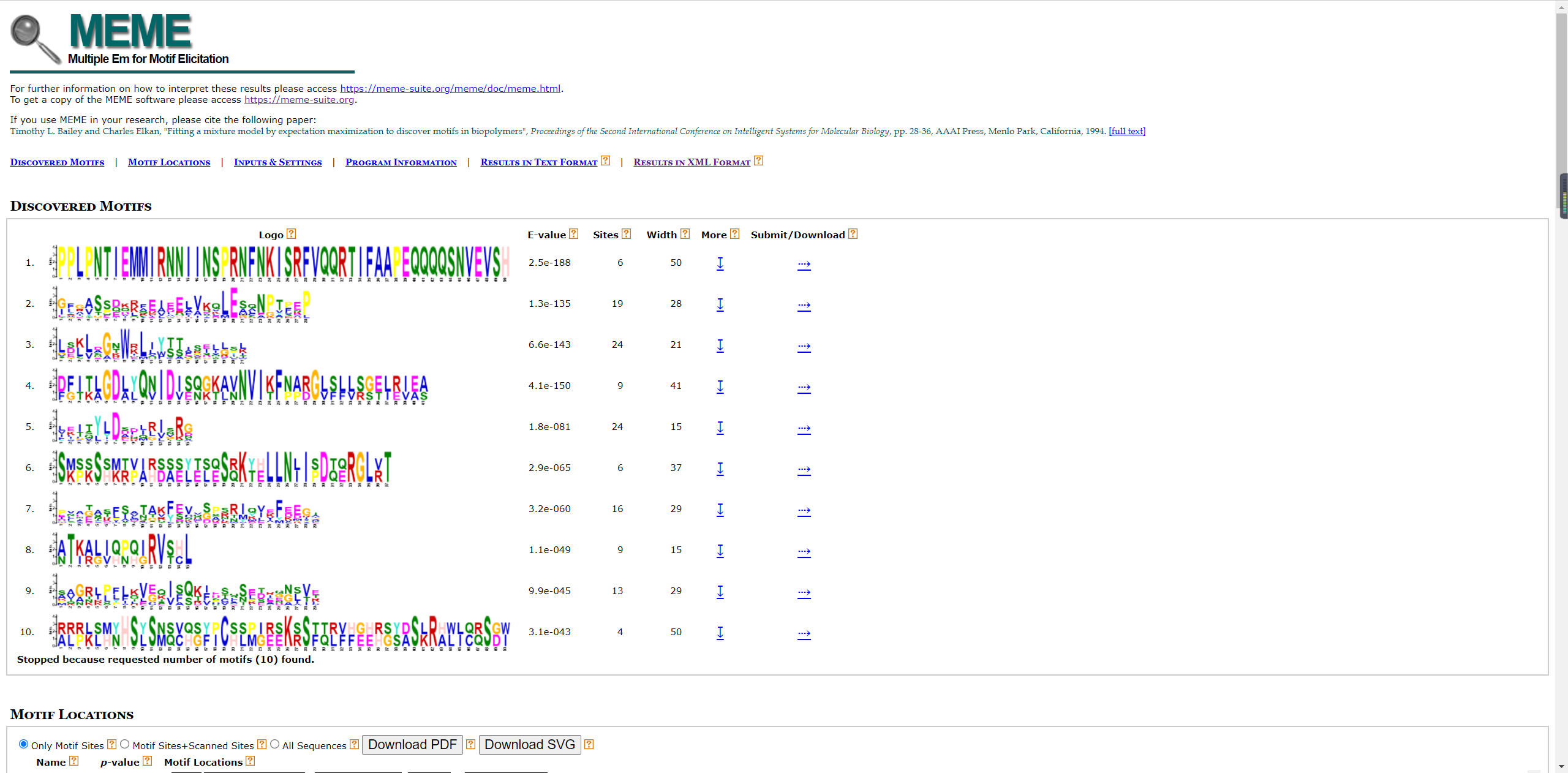 结果文件中就有seqlogo文件信息。 也可以自己的下载后绘制。
结果文件中就有seqlogo文件信息。 也可以自己的下载后绘制。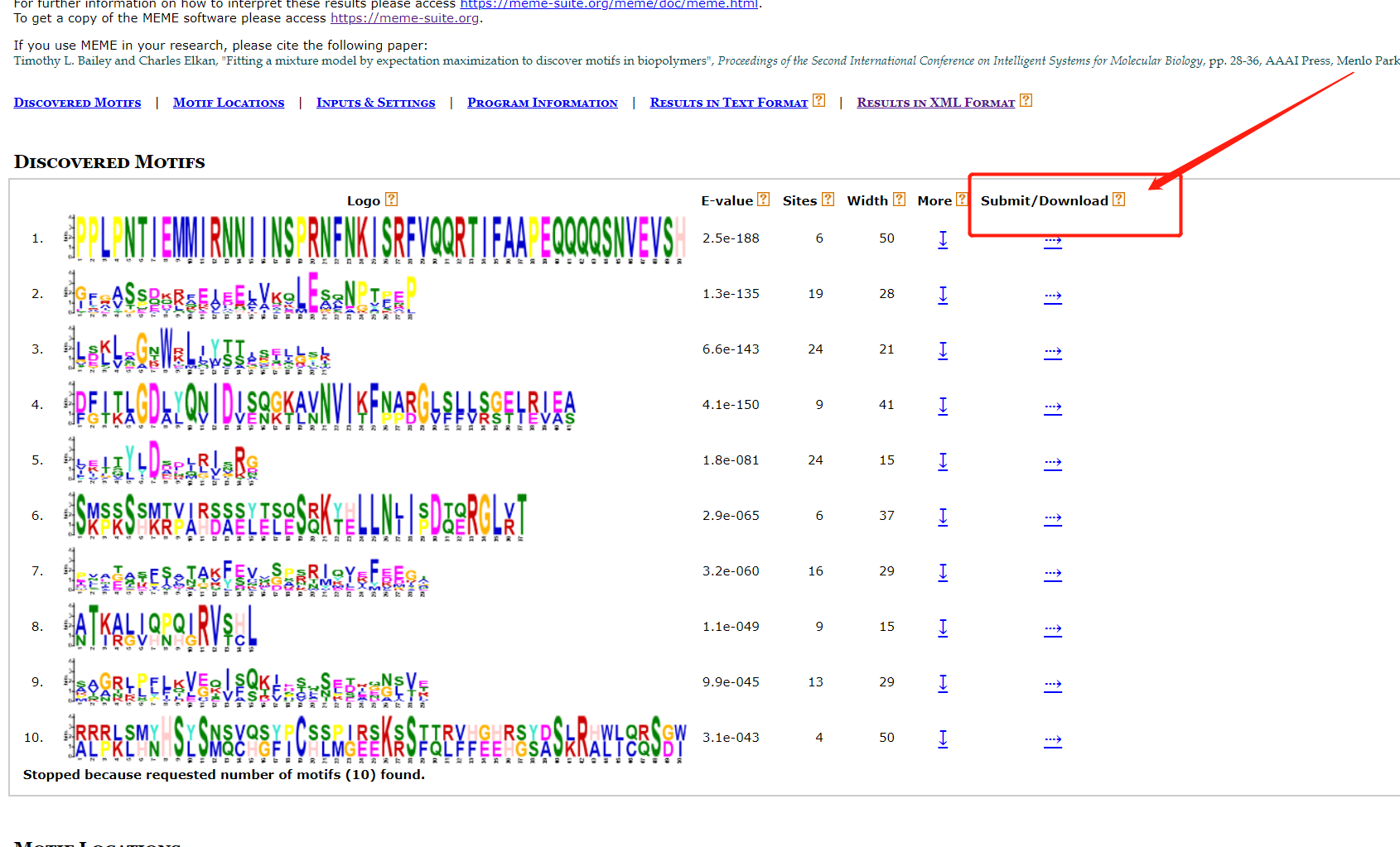 按以下操作即可下载序列。
按以下操作即可下载序列。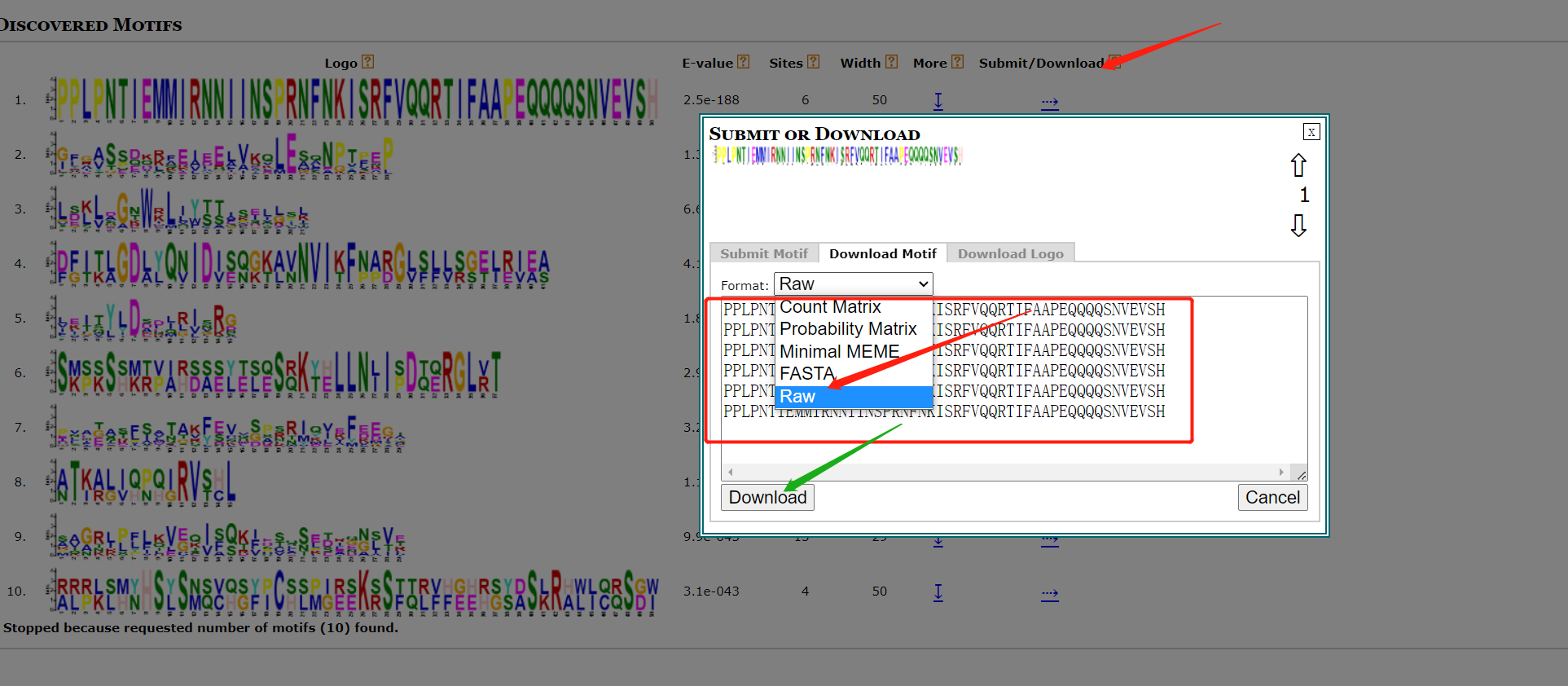 也可以下载已有的seqlogo图片。
也可以下载已有的seqlogo图片。 下载后所有的motif序列信息。
下载后所有的motif序列信息。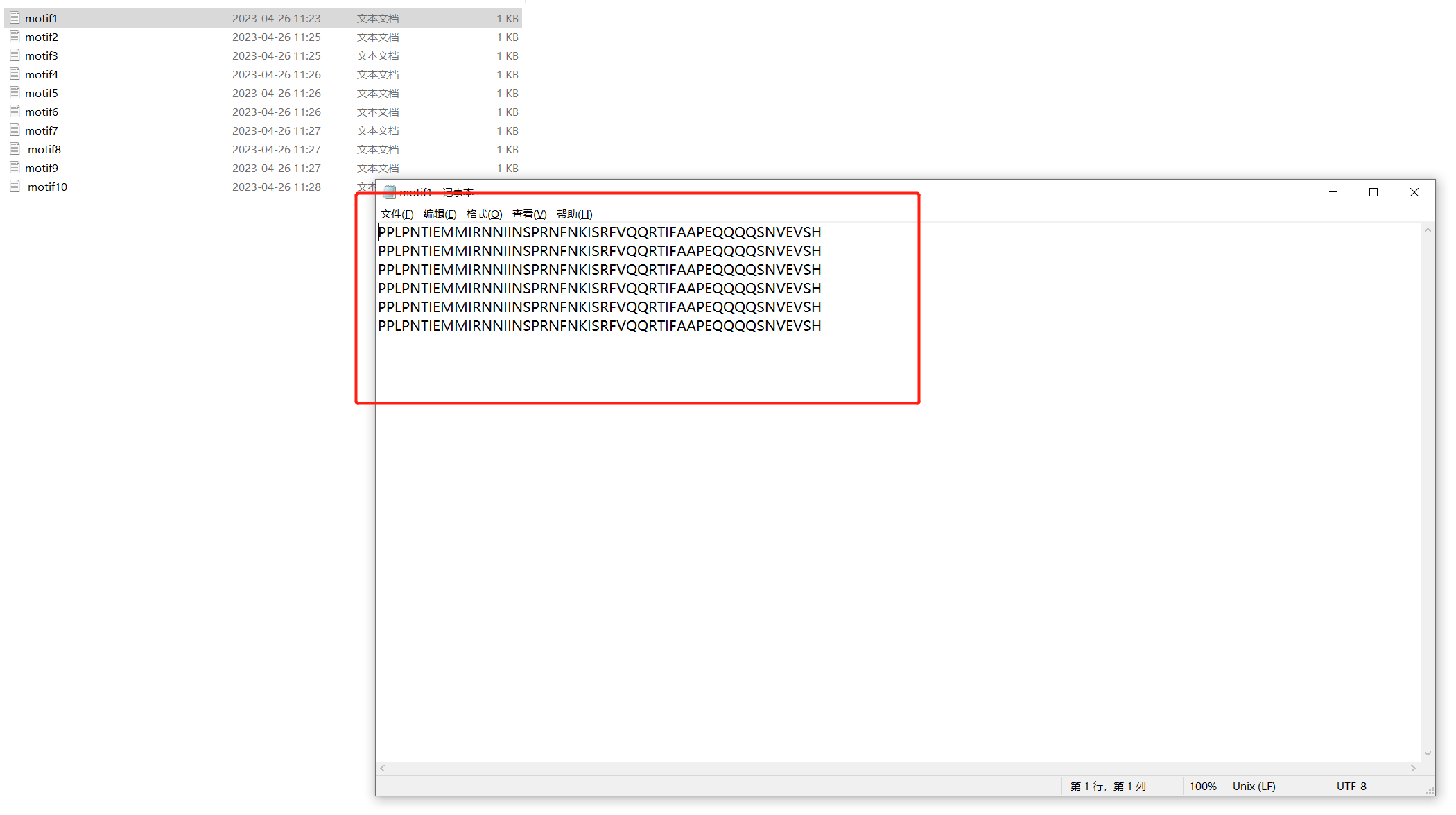
- 使用R语言对Seqlogo序列进行可视化 这里借用这篇教程,基因结构及motif分析。批量生产Seqlogo可视化。
我们可以根据自己的motifi数量进行命名,我自己只有10个motif信息。所以命名为motif1-10.txt。
## 加载所需要的包
library(ggplot2)
#BiocManager::install("ggseqlogo")
library(ggseqlogo)
## 批量生产文件名
filelist = c(paste0('motif',1:10,'.txt'))
filelen <- length(filelist)
##批量读取
data.list <- list()
for (i in 1:filelen) {
data.list[[paste0('motif',i)]]=scan(filelist[i],what = '')
}
ggseqlogo(data.list,col_scheme="clustalx", ncol = 5)+
theme(axis.line = element_line(colour = 'black'),
axis.text.x = element_blank(),
legend.title = element_blank())
ggplot()+
geom_logo(data.list, col_scheme = "clustalx")+
theme_logo()+
facet_wrap(~seq_group,ncol = 5,scales = "free_x")+
theme(axis.line = element_line(colour = 'black'),
axis.text.x = element_blank())
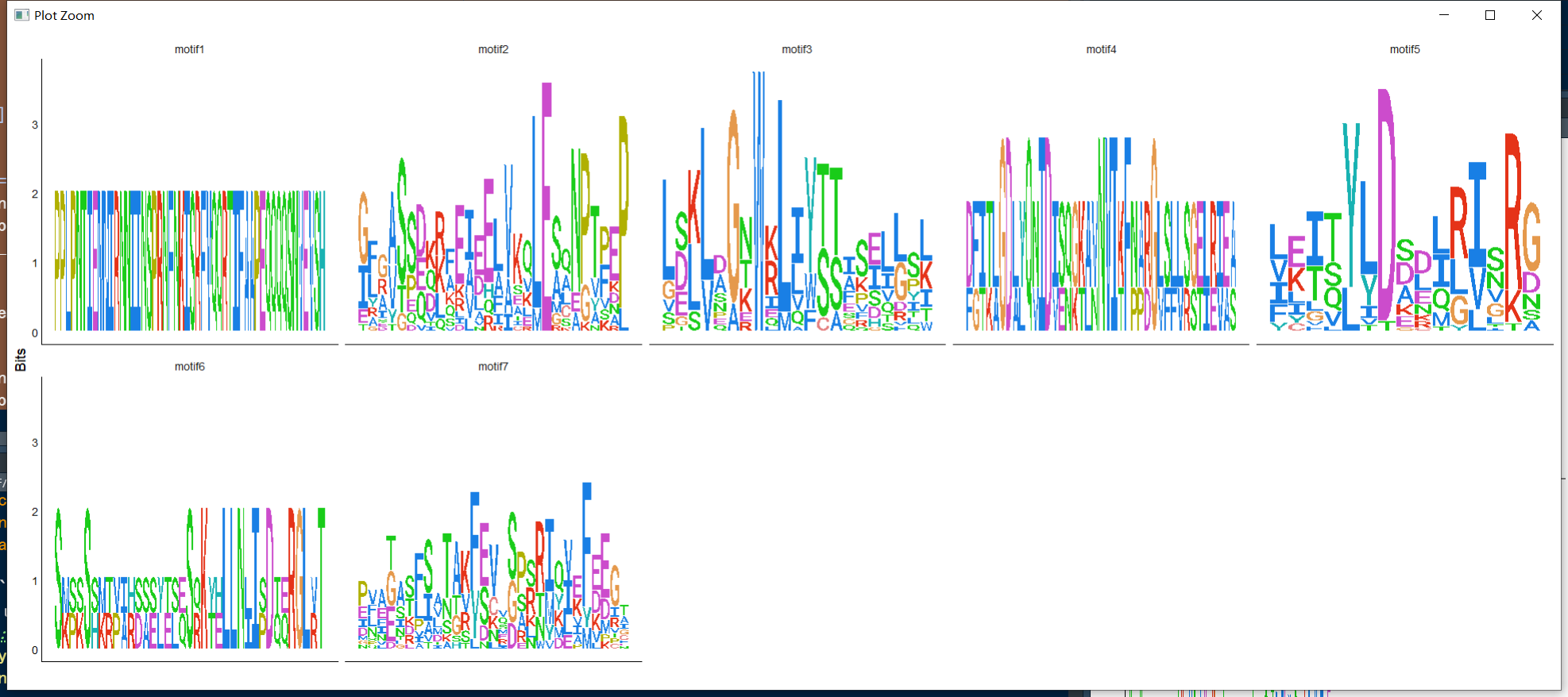 对比一下MEME网站中的图形。
对比一下MEME网站中的图形。 对于Seqlogo的绘制,美化,可以根据很多优秀的教程。在网上上一搜,都可以找到。
对于Seqlogo的绘制,美化,可以根据很多优秀的教程。在网上上一搜,都可以找到。
2.4.3 motif的分析
- 下载结果文件MAST XML output,使用TBtools软件进行可视化。
- 打开TBtools中的Gene Structure View,只需上传MEME中的XML文件即可,上传上去直接点击Start。
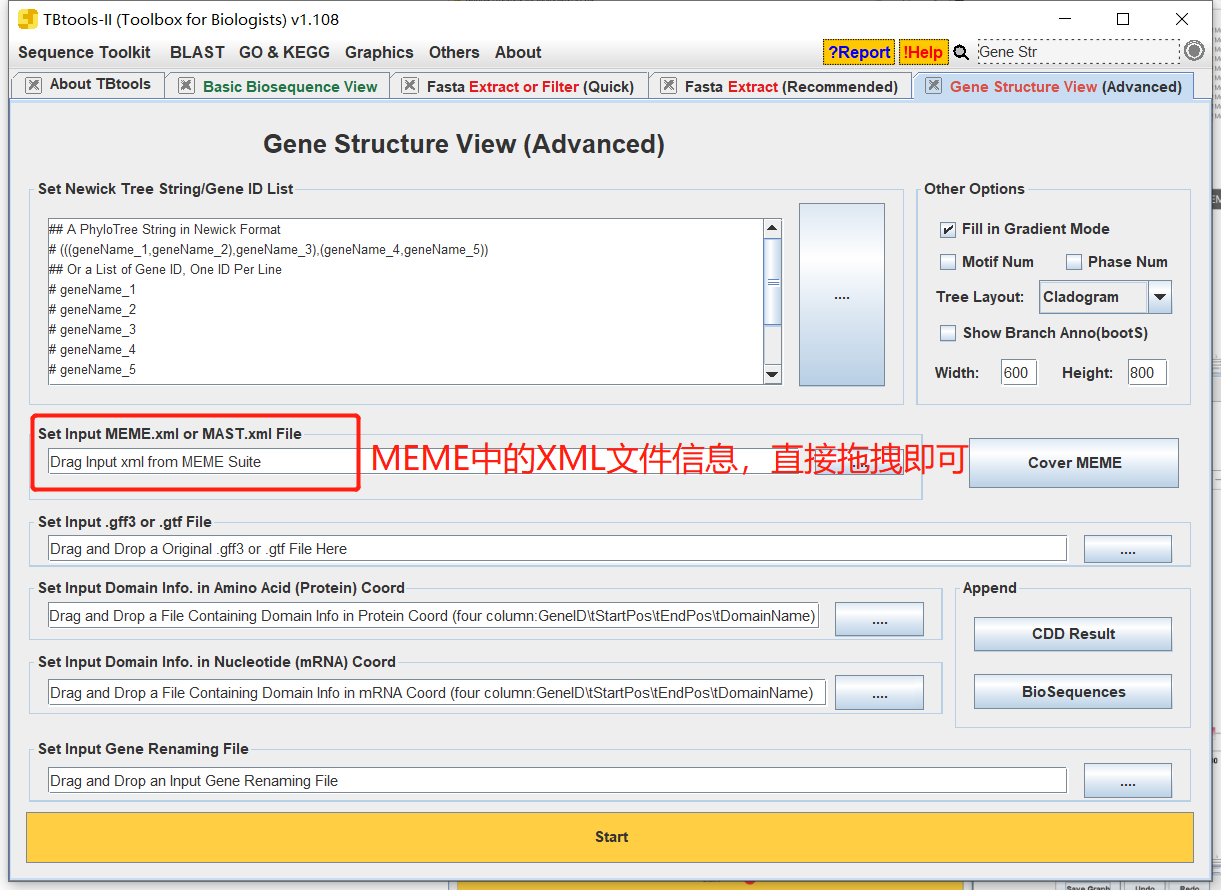 -- 操作:
-- 操作: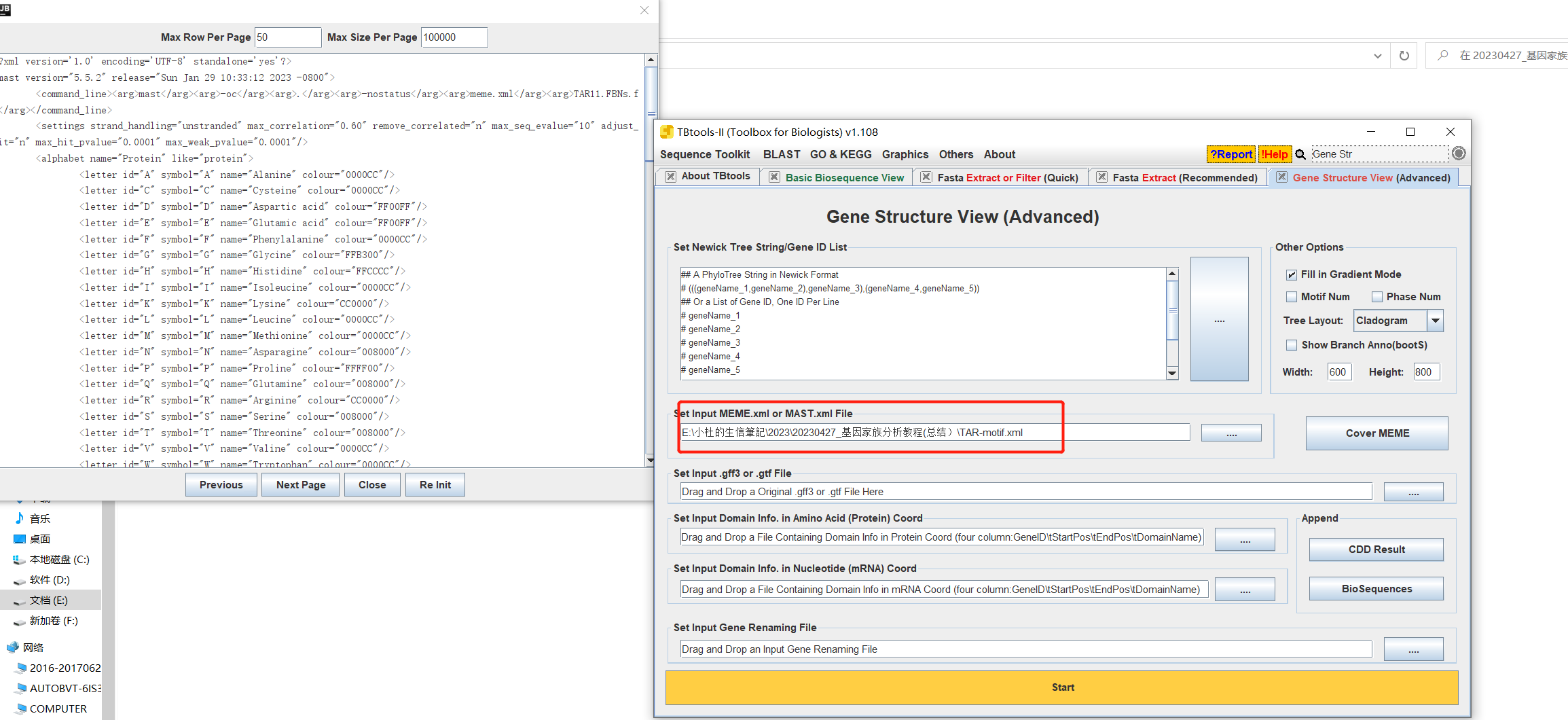 结果:
结果: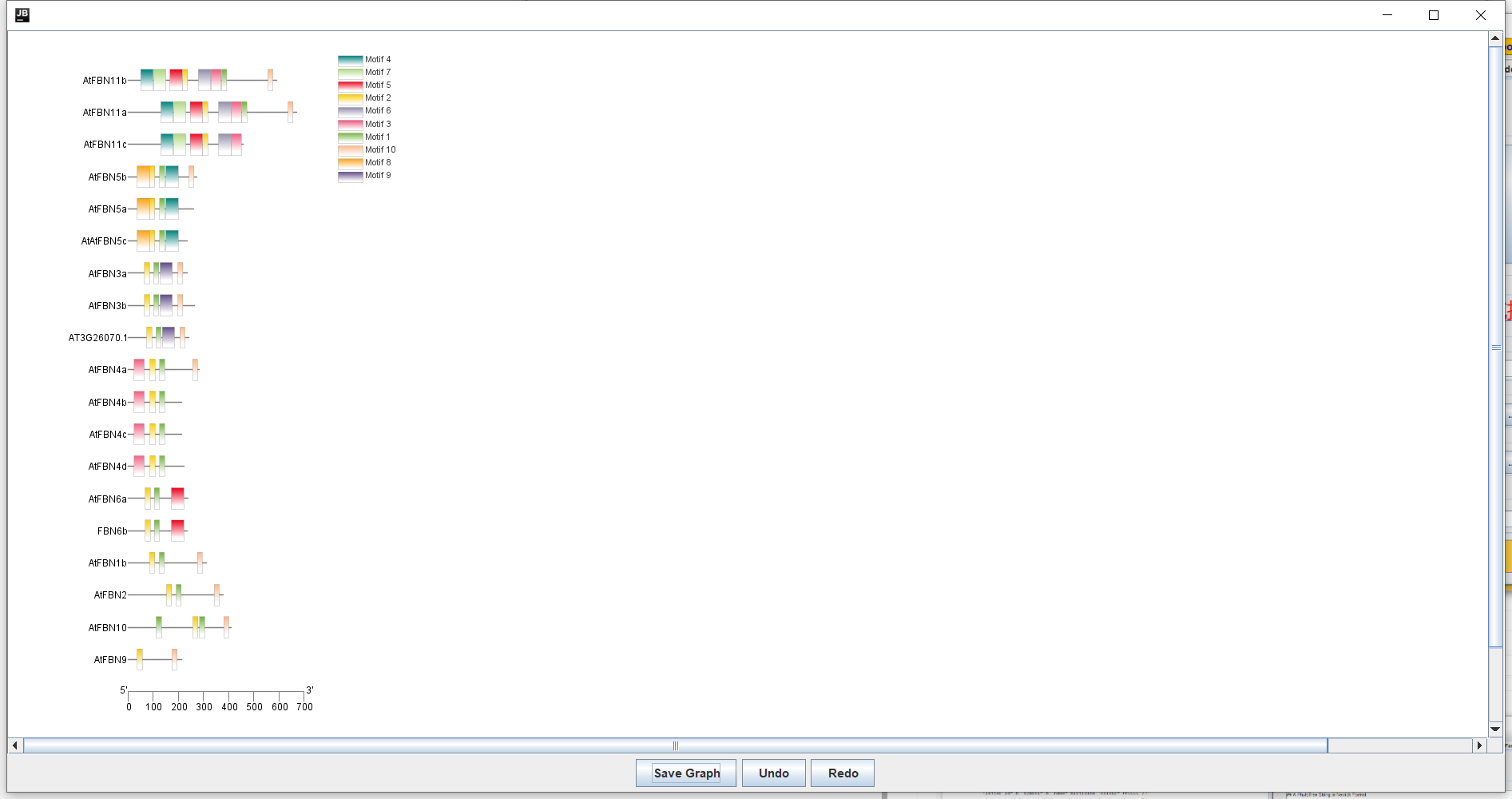 保存!!
保存!!
注意:我们这里保存的时候最好保存为PDF或SVG格式,输出为矢量图**。**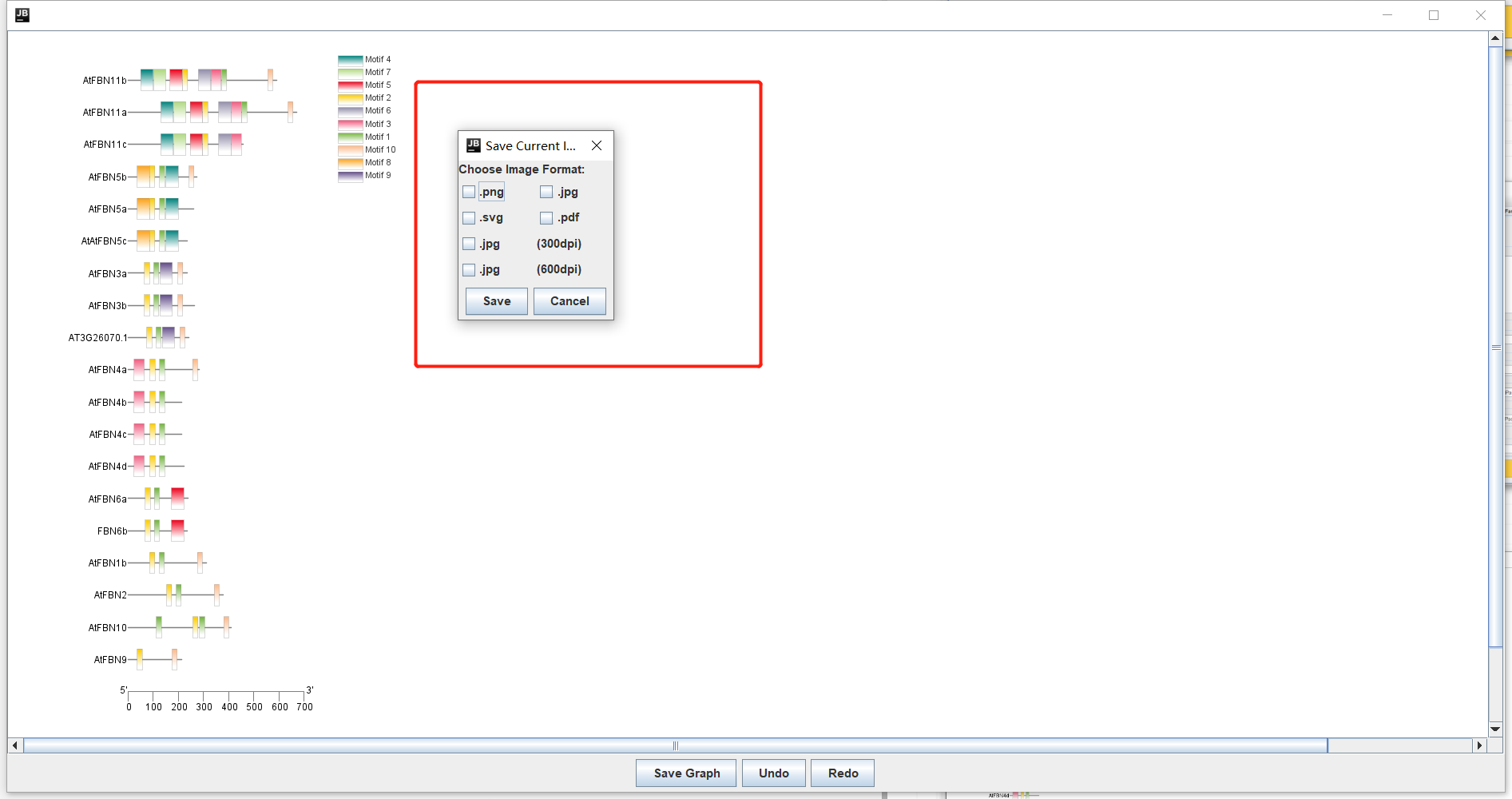
如果我们的教程只是到这里,那么就没有什么意义了。因为,类似非常优秀和详细的教程很多。绘制出图形是一方面、美化可是重头戏**。**
在MEME输出文件中,也提供了motif的图形,也可直接使用。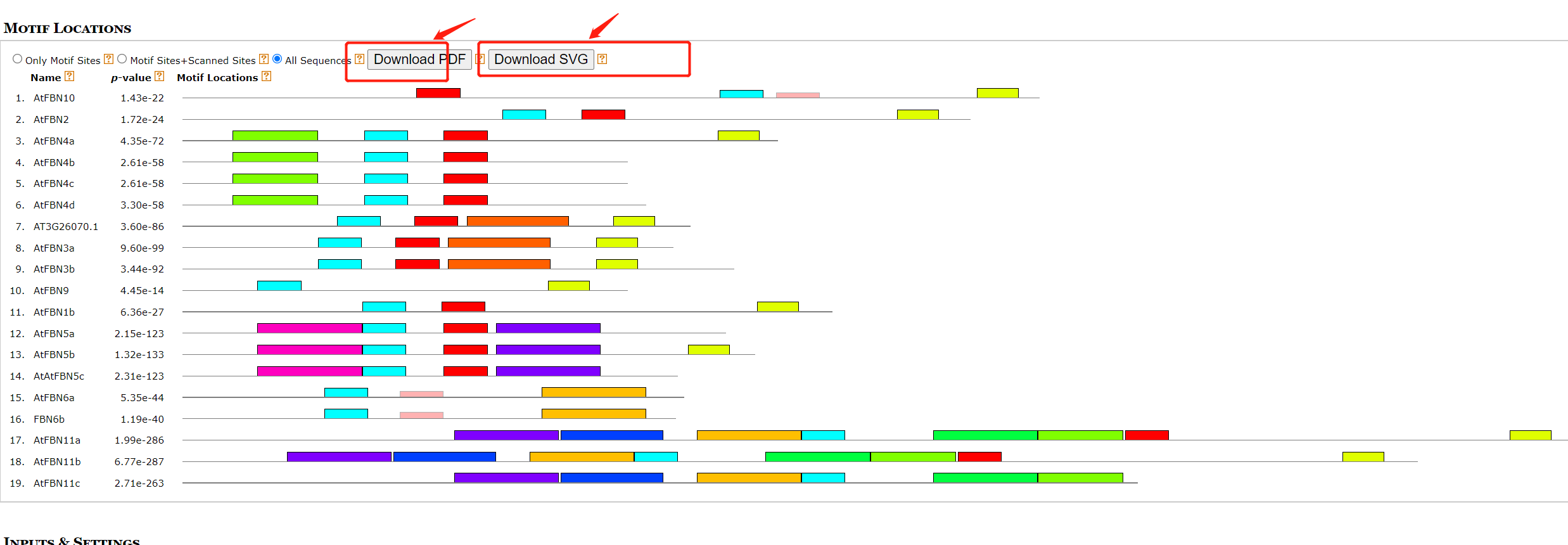
2.5 基因家族保守结构域分析
- 使用Batch CD-Search进行预测,网址:https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi

- 提交序列信息即可
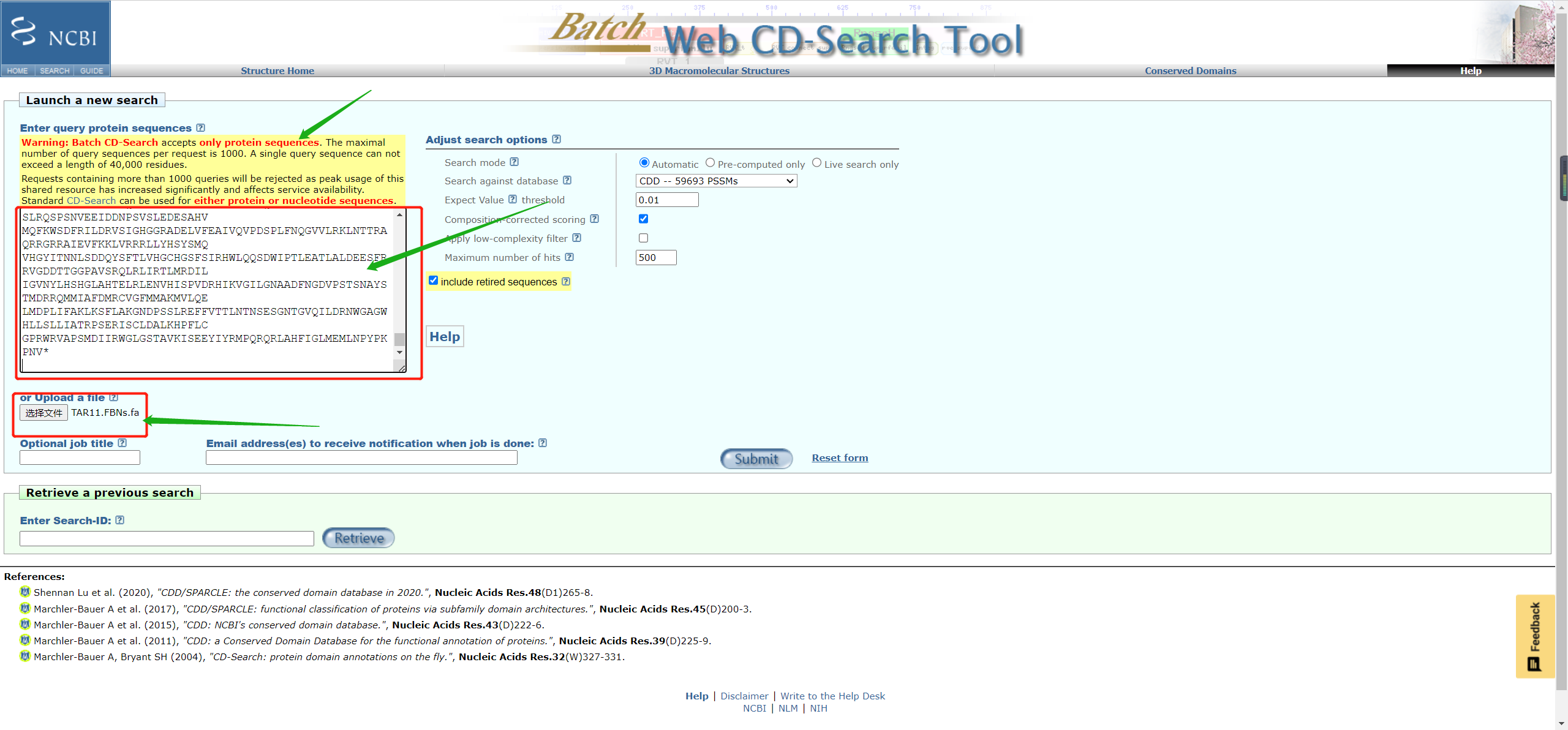
- Batch CD-search只支持目的基因**蛋白序列**信息, 以及序列数量少于1000。
Warning: Batch CD-Search accepts only protein sequences**. The maximal number of query sequences per request is 1000**. A single query sequence can not exceed a length of 40,000 residues.
可以提供你的邮箱,等运行结束后,直接发送到你的邮箱。如果序列较多,建议提供邮箱。
- 下载文件
 结果文件:
结果文件: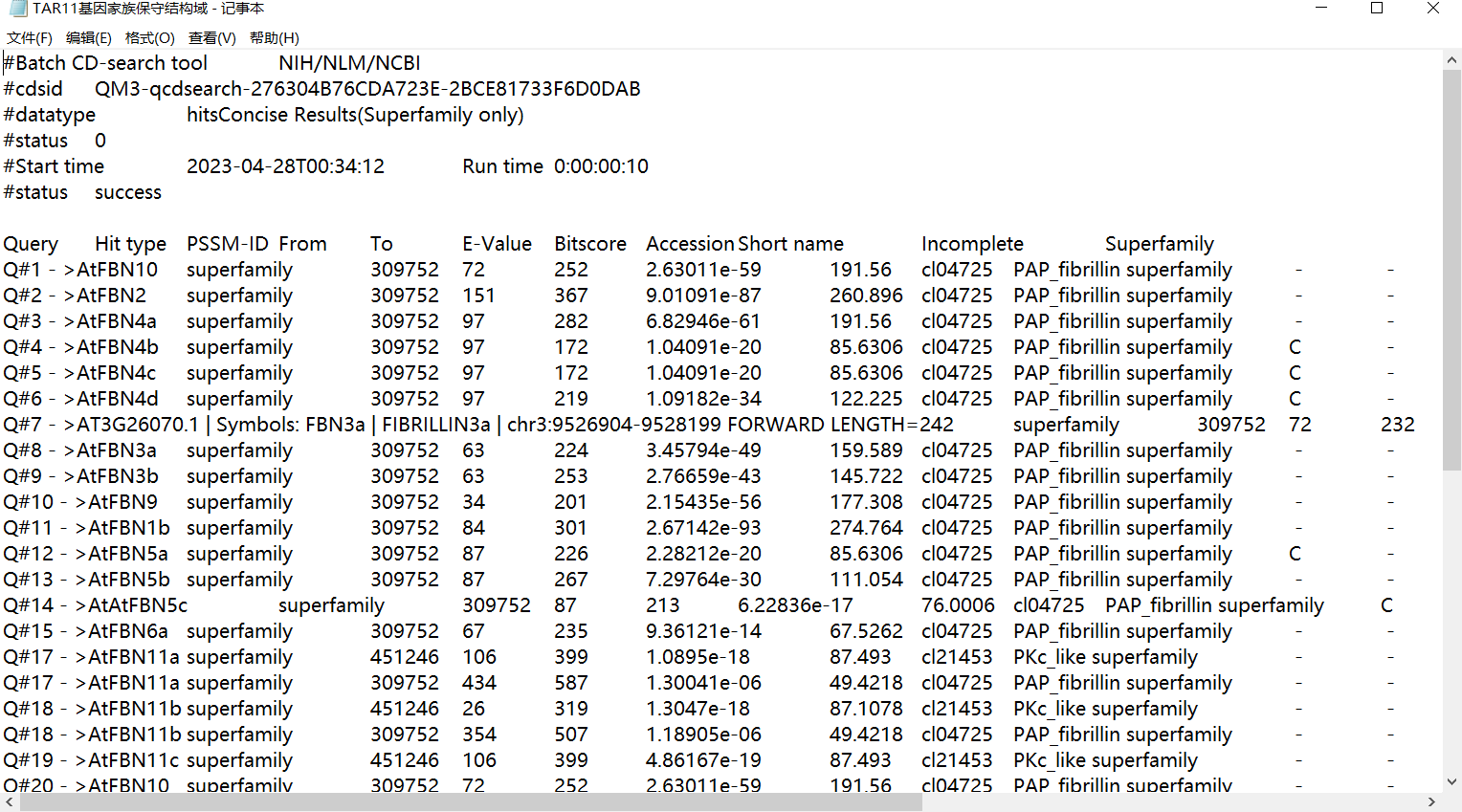
- 打开TBtools中的Visualize NCBI CDD DOmainPattern
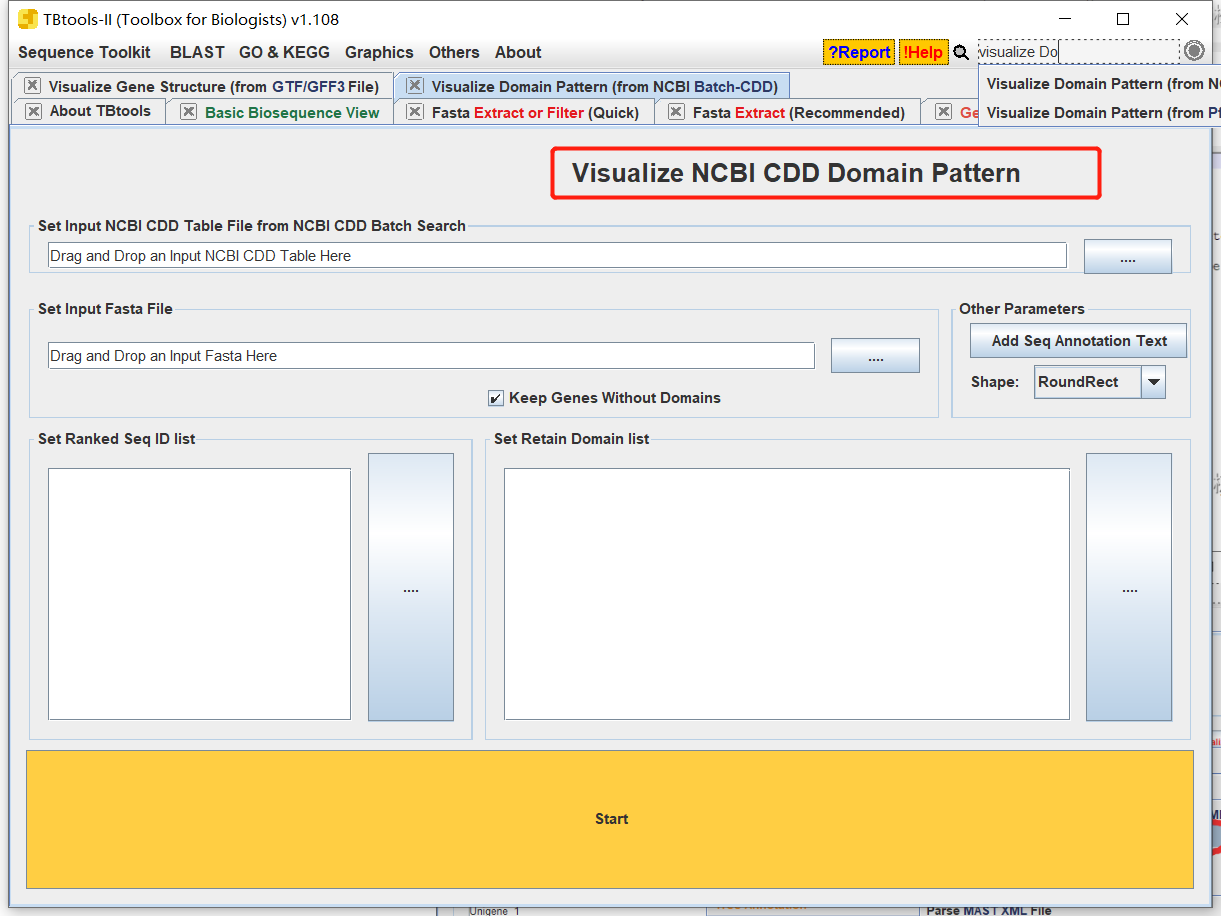
- 输入结果文件和fa文件
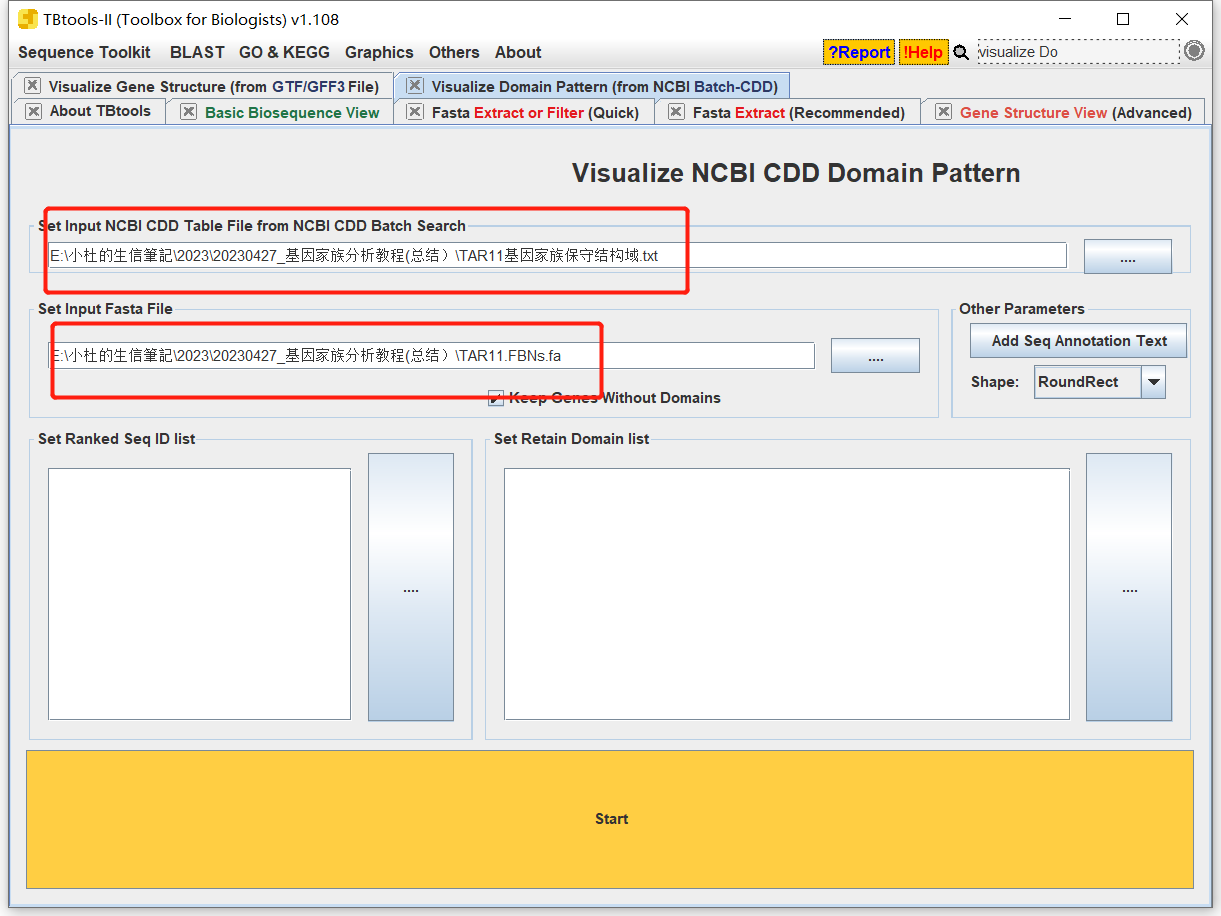
- 根据自己的需求进行调整即可。
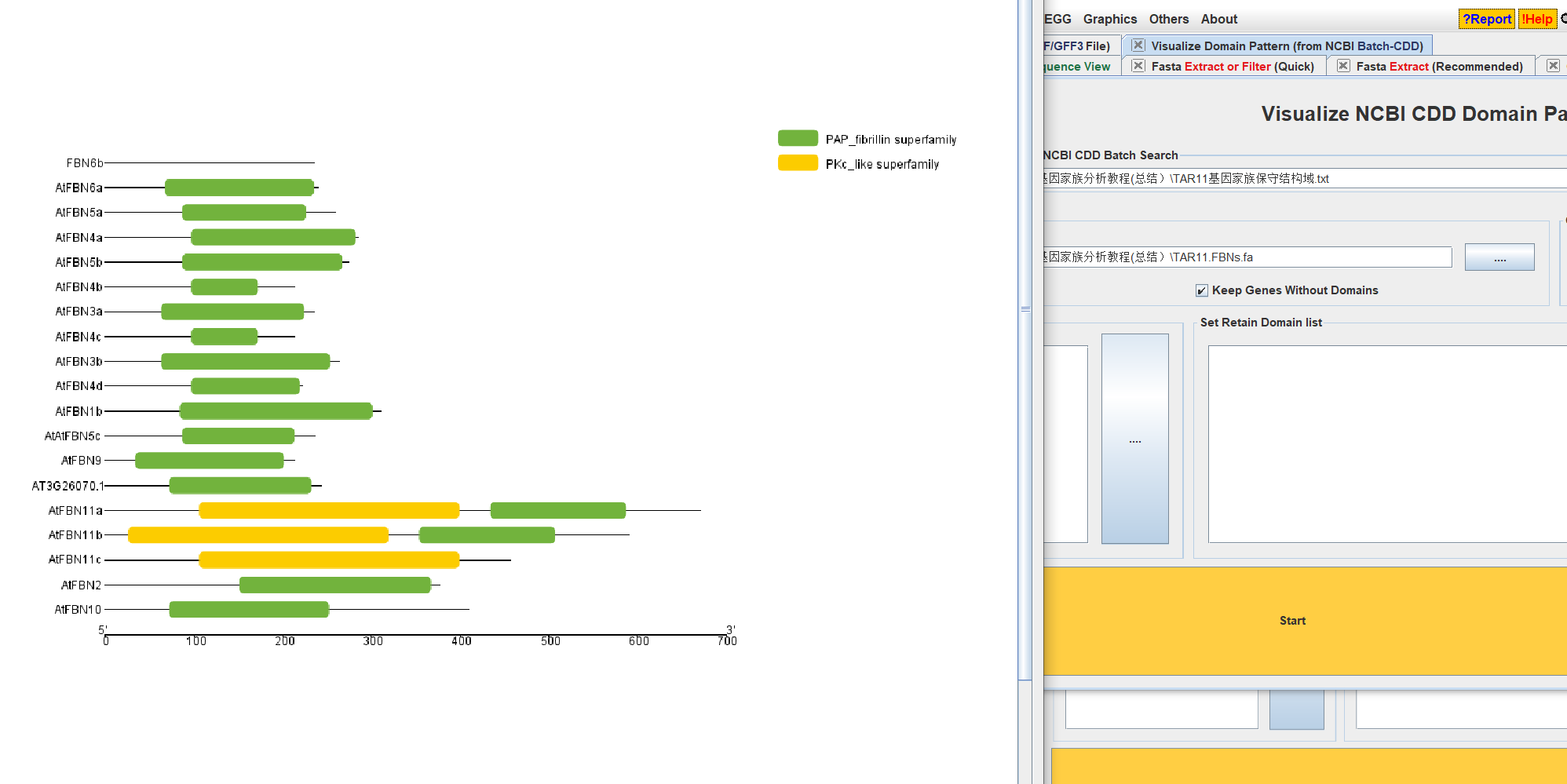
- 输出文件。
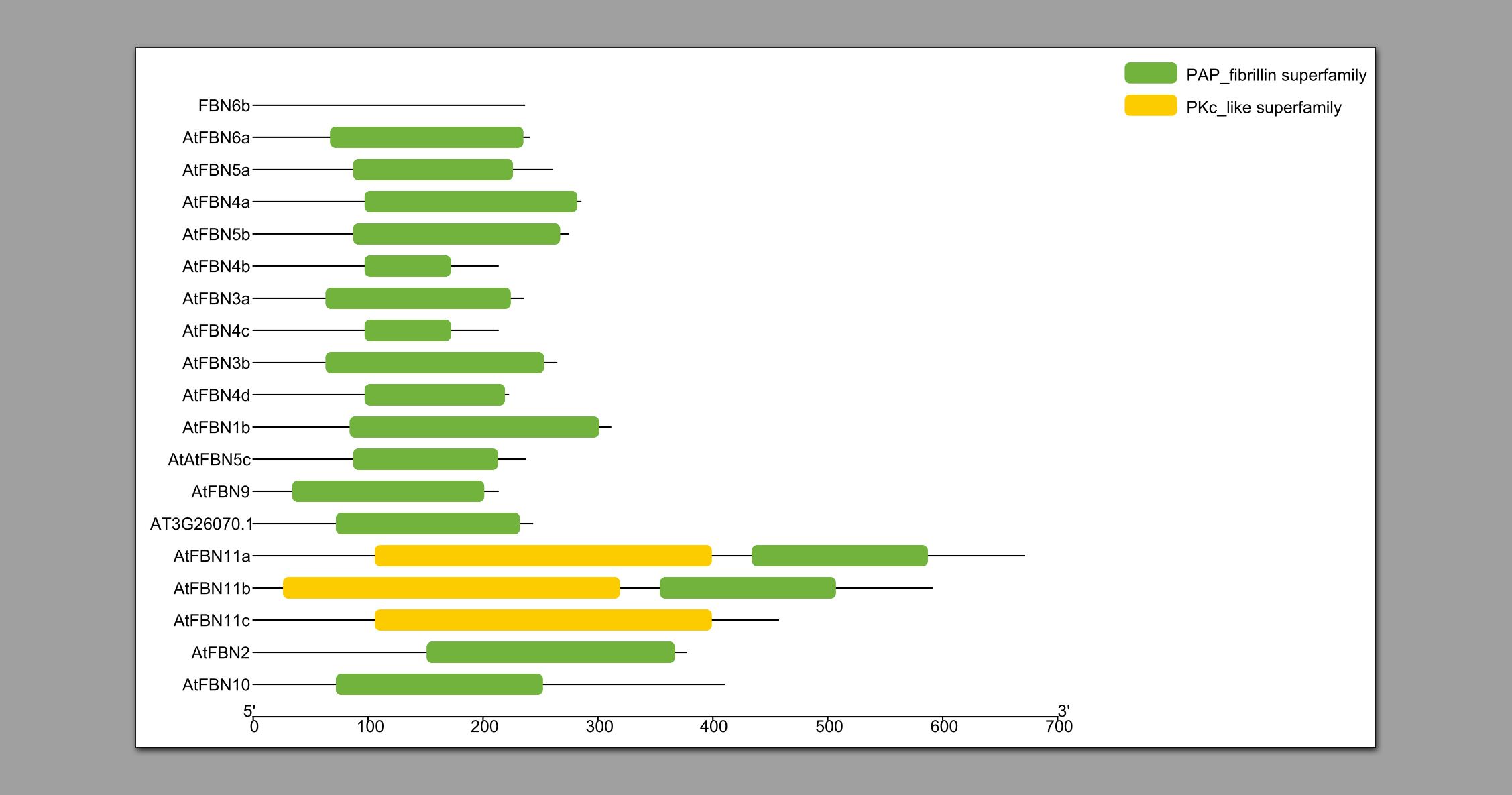
2.6 进化树分析
进化树分析,在基因家族中是必须的,以及在很多图中都是需要的。进化树分析和绘制,也有很多教程,参考iqtree+ggtree绘制进化树教程、或是你也可以使用MEGA来做分析。
2.6.1 iqtree+ggtree绘制进化树教程
参考**:**iqtree+ggtree绘制进化树教程
- iqtree获得树文件
所需软件
- mafft
- iqtree mafft安装 我是使用服务器中运行的,安装可以使用conda
conda install mafft
iqtree官网
http://www.iqtree.org/
 iqtree功能很强大,大家可以查看软件的官方文档。 **安装**
iqtree功能很强大,大家可以查看软件的官方文档。 **安装**
conda install iqtree
 软件安装好后直接运行即可。
软件安装好后直接运行即可。
- 序列准备
进化树序列可以使用蛋白序列或核酸序列即可,格式按其准备即可。
>B2LU34
MTSIAFWNAFTVNPFPAAARRSPPPLTPFTSGALSPARKPRILEISHPRTLPSFRVQAIAEDEWESEKKALKGVVGSVAL
AEDETTGADLVVSDLKKKLIDQLFGTDRGLKATSETRAEVNELITQLEAKNPNPAPTEALSLLNGRWILAYTSFAGLFPL
LGAESLQQLLKVDEISQTIDSEGFTVQNSVRFVGPFSSTSVTTNAKFEVRSPKRVQIKFEEGIIGTPQLTDSIVIPDKFE
FFGQNIDLSPFKGVISSLQDTASSVAKTISSQPPIKFPISNSNAQSWLLTTYLDDELRISRADGGSVFVLIKEGSPLLT
>B4F6G1
MTSIAFCNAFTVNPFLAAARRSPPPLTPLTSVALSPARKPRILAIFHPRTFPSFRVQAIAEDEWESEKKTLKGVVGSVAL
AEDEKTGADLVVSDLKKKLIDQLFGTDRGLKATSETRAEVNELITQLEAKNPNPAPTEALSLLNGKWILAYTSFVGLFPL
LGAESLQQLLKVDEISQTIDSEGFTVQNSVRFVGPFSSTSVTTNAKFEVRSPKRVQIKFEEGIIGTPQLTDSIVIPDKVE
FFGQNIDLSPFKGVISSLQDTASSVAKTISSQPPIKFPISNSNAQSWLLTTYLDDELRISRADGGSVFVLILESSPLLT
>O49629
MATVQLSTQFSCQTRVSISPNSKSISKPPFLVPVTSIIHRPMISTGGIAVSPRRVFKVRATDTGEIGSALLAAEEAIEDV
EETERLKRSLVDSLYGTDRGLSASSETRAEIGDLITQLESKNPTPAPTEALFLLNGKWILAYTSFVNLFPLLSRGIVPLI
KVDEISQTIDSDNFTVQNSVRFAGPLGTNSISTNAKFEIRSPKRVQIKFEQGVIGTPQLTDSIEIPEYVEVLGQKIDLNP
IRGLLTSVQDTASSVARTISSQPPLKFSLPADNAQSWLLTTYLDKDIRISRGDGGSVFVLIKEGSPLLNP
- mafft比对
使用mafft将序列对齐。
mafft test.fa > test.aligend.fa
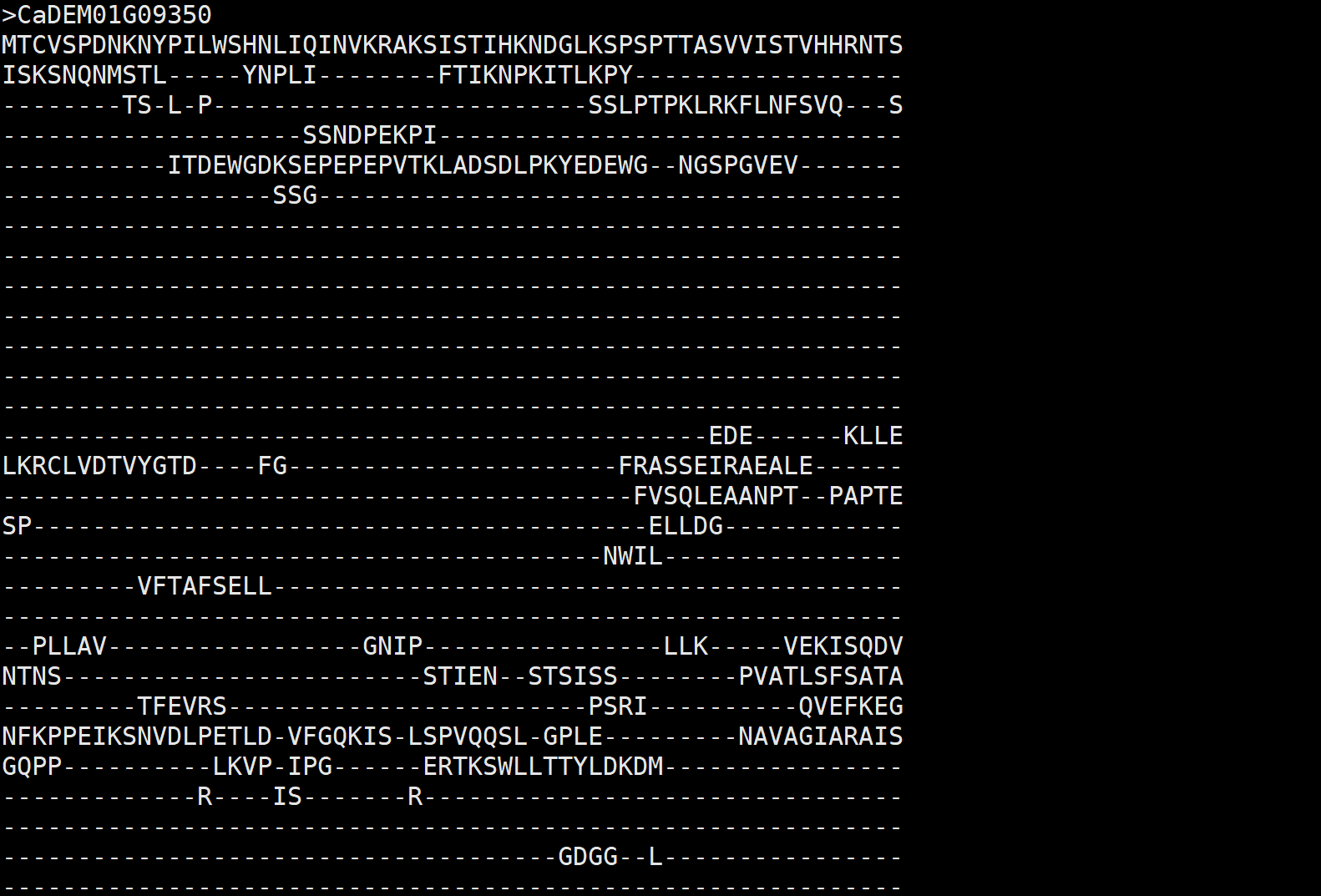 我们获得对齐后的数据格式。
我们获得对齐后的数据格式。
- iqtree构建树
iqtree -s test.aligend.fa -m MFP -bnni -nt AUTO -cmax 15 -redo -bb 1000
关于iqtree的使用,可以看这篇教程IQ-TREE的使用 - 超快速用极大似然法构建进化树,讲的很详细。
必须参数:
-s 输入多序列比对文件
-nt 多线程,AUTO是自动多线程
-bb 1000 指定了要用快速BS法做1000次
最终,我们可以获得以下结果文件。
- ggtree绘制进化树
这里,我们使用基迪奥的教程,如何绘制添加分类色块的进化树?,这个教程也是讲解得很详细。
注意:我们这里使用iqtree输出文件test.aligend.fa.treefile作为输入文件。
#载入相关的R包;
library(ggtree)
library(treeio)
library(ggplot2)
#读入newick格式的进化树文件;
tr = read.newick("test.aligend.fa.treefile")
ggtree(tr)
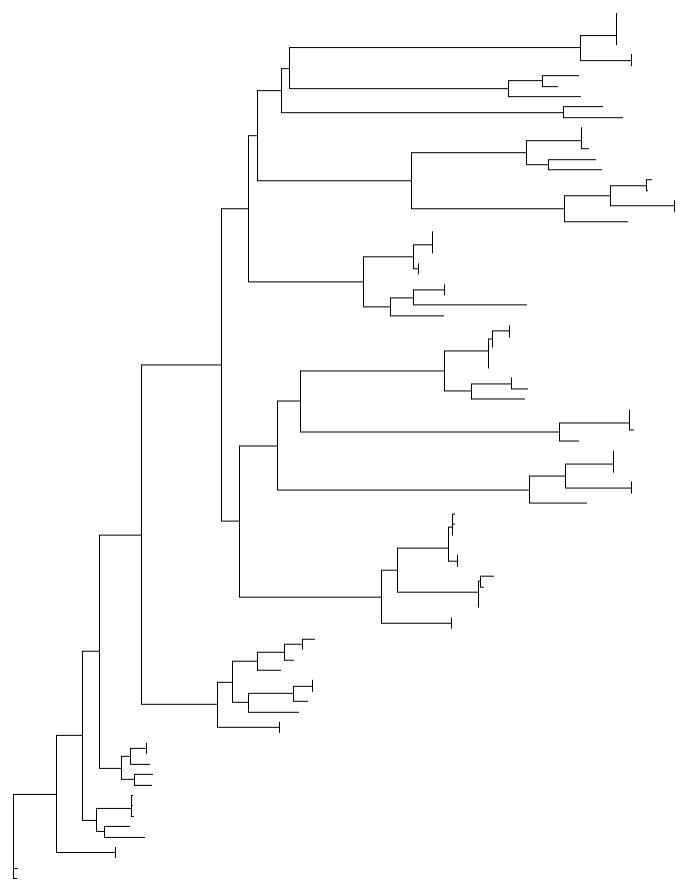
#为进化树添加叶标签;
p1 <- p0 + geom_tiplab(size=2,color="grey10")
p1
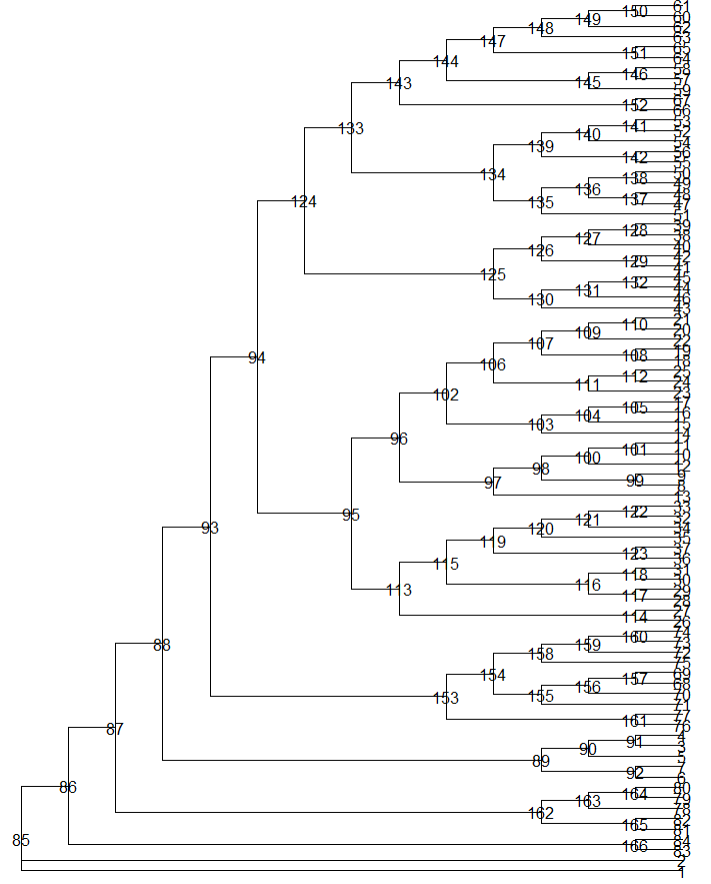
#为进化树添加圆形顶点;
p2 <- p0+ geom_tiplab(size=2,offset=0.03, color="grey10")+
geom_tippoint(color="#6bc72b",fill="#6bc72b",
alpha=0.4, size=3,shape=21)
p2
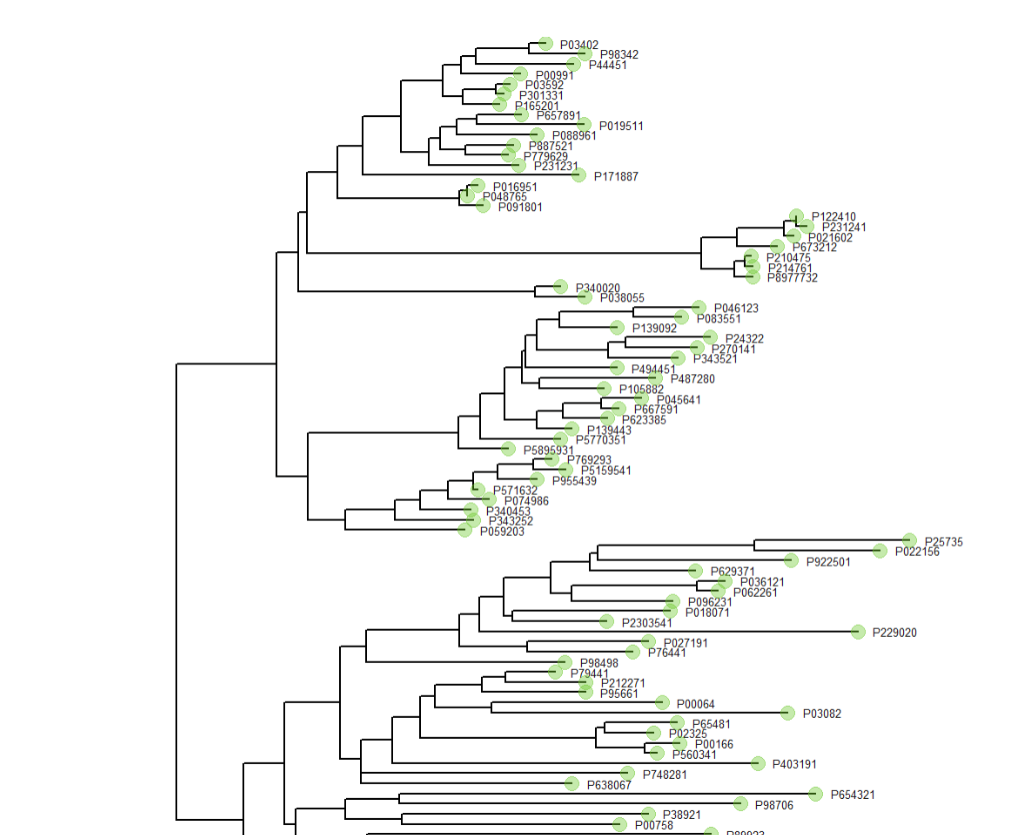
后面的教程参数调整,按着教程即可如何绘制添加分类色块的进化树?
2.6.2 MEGA制作进化树
此部分内容来自:TBtools | 基因家族分析 (进化树、Motifs、结构域) 输入数据为目标基因家族的蛋白质序列。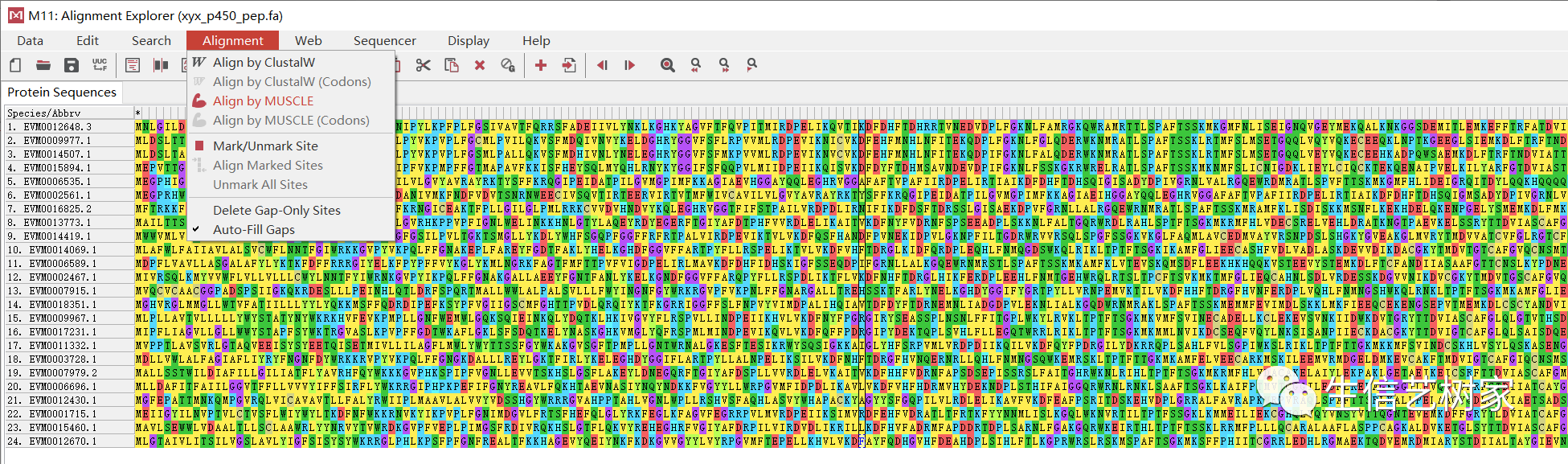
先进行多序列比对,用MUSCLE默认参数。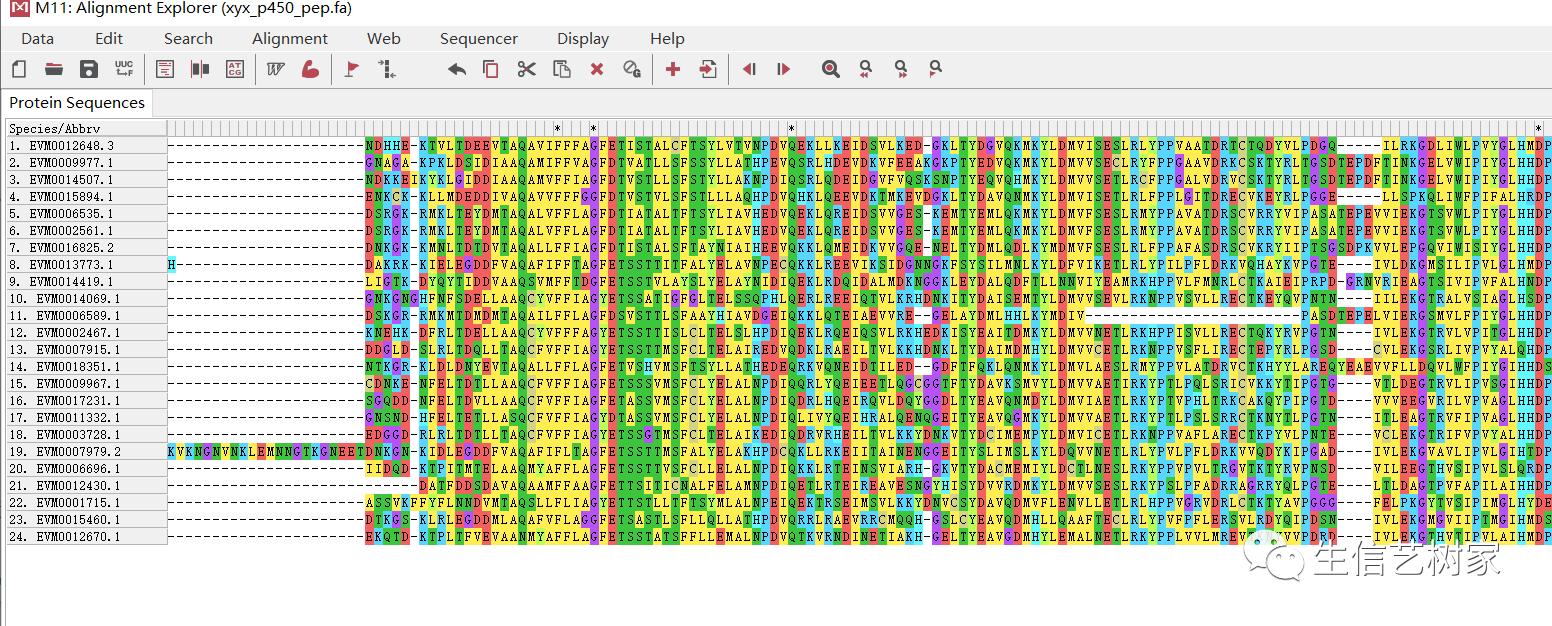 图片将比对好的结果保存为.meg格式。
图片将比对好的结果保存为.meg格式。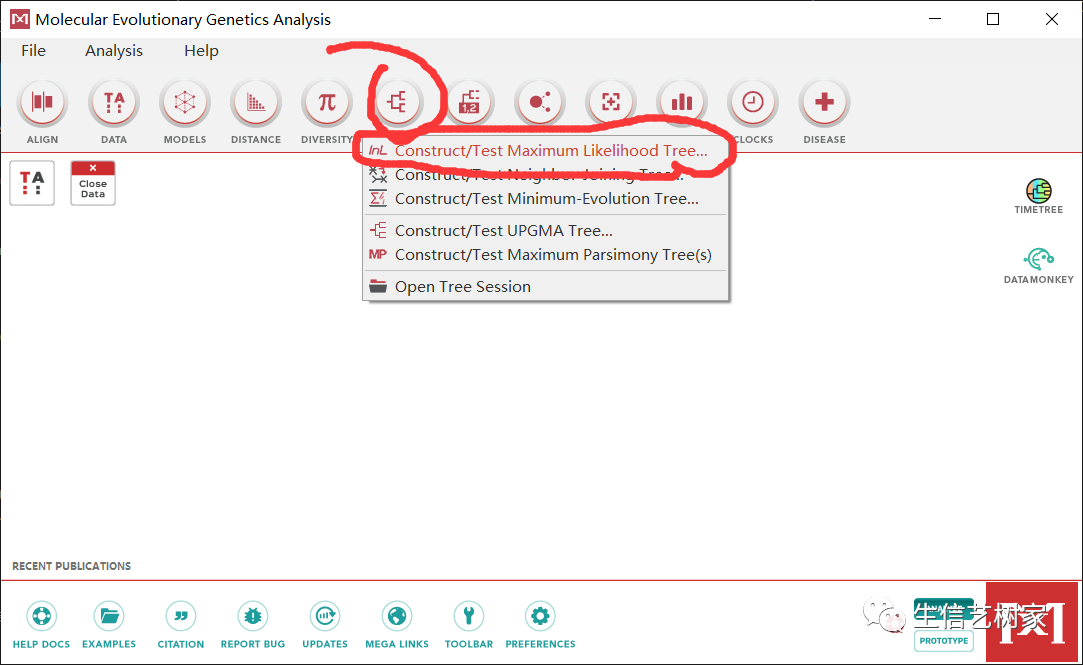 重新打开比对后的文件,构建进化树,使用最大似然法,根据需要选择建树方法。再构建之前可以进行模型的预测,这里节省时间直接使用默认参数。
重新打开比对后的文件,构建进化树,使用最大似然法,根据需要选择建树方法。再构建之前可以进行模型的预测,这里节省时间直接使用默认参数。
现在就构建好了一棵进化树,导出为.nwk格式。接下来最后一步就是再TBtools中展示所有结果。
..................................后续教程还有........................................................................................
若我们的教程对你有所帮助,请点赞+收藏+转发,这是对我们最大的支持。
往期部分文章
「1. 最全WGCNA教程(替换数据即可出全部结果与图形)」
「2. 精美图形绘制教程」
「3. 转录组分析教程」
「4. 转录组下游分析」
「小杜的生信筆記」 ,主要发表或收录生物信息学教程,以及基于R分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!






 苏公网安备32011502012024号
苏公网安备32011502012024号任意下载一个版本即可,安装步骤不再做说明](http://www.hmmer.org/**任意下载一个版本即可,安装步骤不再做说明){kind=link}