用vcf文件和表型关联做GWAS分析的时候通常会要求vcf文件里的样本顺序和表型样本顺序是一致的,一种做法是根据vcf文件里的样本顺序调整表型数据里的样本顺序,另外一种做法是根据表型数据里的样本顺序去调整vcf文件的样本顺序。
调整vcf文件的样本顺序没有找到现成的工具可以做这个事情,之前搜索找到一个链接说bcftools 的 reheader命令可以调整样本顺序,我自己之前也用这个命令做过。最近才发现这个命令不会更改样本的顺序,只是根据提供的样本名把vcf文件里的样本名改掉,基因型的顺序并不会动
比如vcf文件
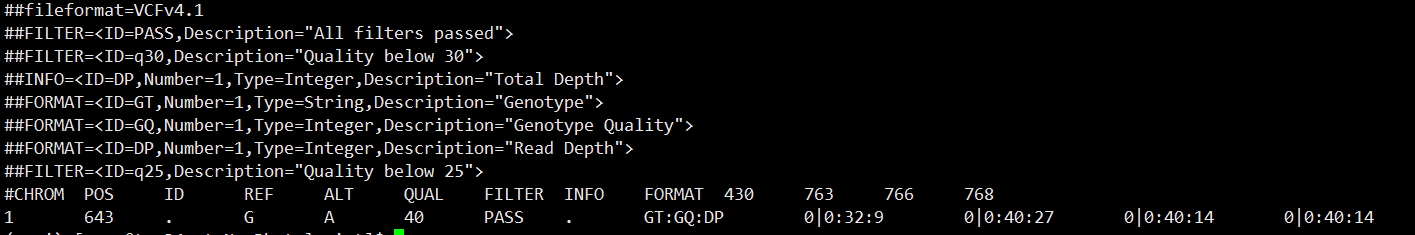
运行bcftools reheader
bcftools reheader -s sample.order practice.recode.vcf
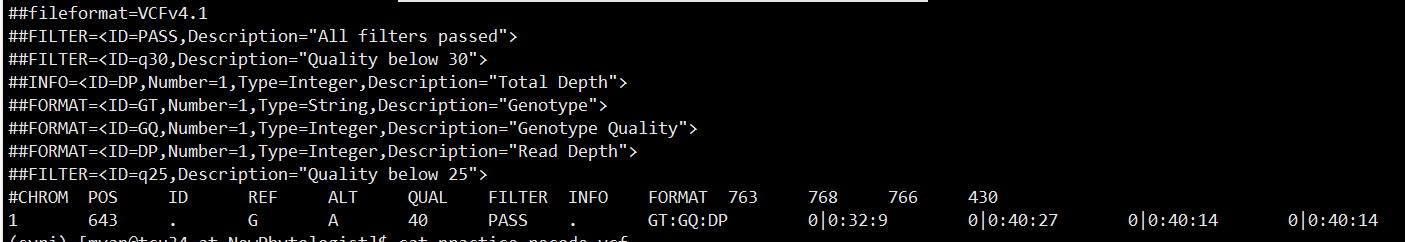
自己写的python脚本
import sys
## list1 指定顺序的列表
## list2 需要调整顺序的列表
def find_positions(list1,list2):
positions = []
for element in list1:
position = list2.index(element)
positions.append(position + 9)
return(list(range(9)) + positions)
## sys.argv[1] 样本顺序文件,每行一个样本
## sys.argv[2] 输入vcf
## sys.argv[3] 输出vcf
sample_order_list = []
with open(sys.argv[1],'r') as fr:
for line in fr:
sample_order_list.append(line.strip())
fr = open(sys.argv[2],'r')
fw = open(sys.argv[3],'w')
for line in fr:
if line.startswith("##"):
fw.write(line)
elif line.startswith("#CHROM"):
raw_sample_order = line.strip().split()[9:]
order_list = find_positions(sample_order_list,raw_sample_order)
temp_list = line.strip().split()
new_list = [temp_list[i] for i in order_list]
fw.write("%s\n"%('\t'.join(new_list)))
else:
temp_list = line.strip().split()
new_list = [temp_list[i] for i in order_list]
fw.write("%s\n"%('\t'.join(new_list)))
fr.close()
fw.close()
运行命令
python adjustVCFsampleOrder.py sample.order practice.recode.vcf practice.reorder.vcf
欢迎大家关注我的公众号
小明的数据分析笔记本
小明的数据分析笔记本 公众号 主要分享:1、R语言和python做数据分析和数据可视化的简单小例子;2、园艺植物相关转录组学、基因组学、群体遗传学文献阅读笔记;3、生物信息学入门学习资料及自己的学习笔记!
| 




 苏公网安备32011502012024号
苏公网安备32011502012024号