动动发财的小手,点个赞吧!
1. 基因集检测
转录因子或表观遗传标记可能作用于按共同生物学特征(共享生物学功能、RNAseq 实验中的共同调控等)分组的特定基因组。
ChIPseq 分析中的一个常见步骤是测试常见基因集是否富含转录因子结合或表观遗传标记。
精心策划的基因集的来源包括 GO 联盟(基因的功能、生物过程和细胞定位)、REACTOME(生物途径)和 MsigDB(计算和实验衍生)。
2. ChIPseq 的基因集测试
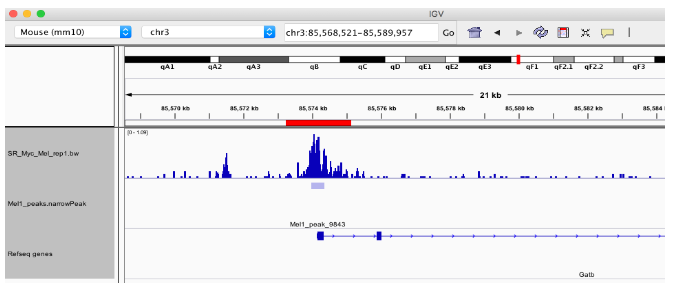
可以对具有与其相关联的峰的基因集执行基因集富集测试。在这个例子中,我们将考虑峰值在基因 TSS 1000bp 以内的基因。
我们不会在测试中直接访问这些数据库库,但会使用广泛使用它们的其他 R/Bioconductor 库。
3. GO 和基因集测试
要在这里执行基因集测试,我们将使用 clusterProfiler 包。clusterProfiler 提供多种富集函数,允许将您的基因列表与已知(例如 GO、KEGG)或自定义基因集进行比较。
在这个例子中,我们使用我们发现与 Myc 峰重叠的所有 TSS 站点。落在 TSS 区域的峰将在我们带注释的 GRanges 对象的注释列中标记为“启动子”。
annotatedPeaksGR[1, ]
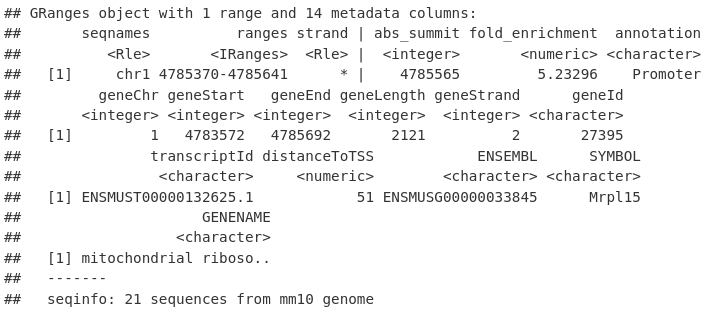
我们可以通过对带注释的 GRanges 进行子集化并从 geneId 列中检索基因名称来提取 TSS 中具有峰的基因的唯一名称。
annotatedPeaksGR_TSS <- annotatedPeaksGR[annotatedPeaksGR$annotation == "Promoter",
]
genesWithPeakInTSS <- unique(annotatedPeaksGR_TSS$geneId)
genesWithPeakInTSS[1:2]

接下来,我们可以提取包含在 TxDb 对象中的所有基因,以用作我们用于通路富集的基因域。
allGeneGR <- genes(TxDb.Mmusculus.UCSC.mm10.knownGene)
allGeneGR[1:2, ]

allGeneIDs <- allGeneGR$gene_id
一旦我们有了相同格式的基因列表和基因域,我们就可以在 enrichGO 函数中使用它们来执行基因本体分析
对于 ont 参数,我们可以在“BP”、“MF”和“CC”子本体之间进行选择,或者为所有三个选择“ALL”。
library(clusterProfiler)
library(org.Mm.eg.db)
GO_result <- enrichGO(gene = genesWithPeakInTSS, universe = allGeneIDs, OrgDb = org.Mm.eg.db,
ont = "BP")
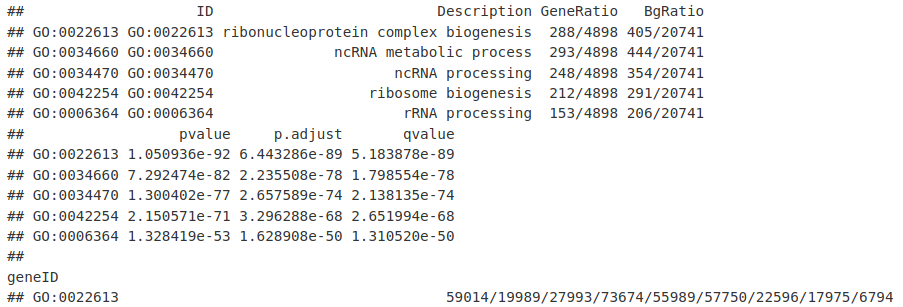
我们现在有一个 enrichResult 实例。从这个对象中,我们可以提取最丰富的基因本体类别的数据框。
GO_result_df <- data.frame(GO_result)
GO_result_df[1:5, ]
可以使用 enrichplot 包从任何 enrichResult 对象生成网络图。
我们测量各种重要基因集之间的相似性并相应地对它们进行分组。 showCategory 参数指定要显示的顶级基因本体命中数。
library(enrichplot)
GO_result_plot <- pairwise_termsim(GO_result)
emapplot(GO_result_plot, showCategory = 20)

除了基因本体之外,我们还可以使用 clusterProfiler enricher 函数针对我们作为 gmt 文件导入的自定义基因集测试我们的基因列表。类似于 enrichGO 函数,这将生成一个可用于可视化的 enrichResult 对象。
在这里,我们将使用 msigdbr 包从 MSigDB 获取基因集。我们还可以运行 msigdbr_collections 函数来查看将用于访问基因集的类别和子类别代码。
library(msigdbr)
msigdbr_collections()

从上一张幻灯片的数据框中,我们可以识别我们想要的类别/子类别,并在 msigdbr 函数中使用它们。这里我们将使用“H”来访问 Hallmark 基因集,最后我们需要得到一个数据框,其中第一列包含基因集的名称,第二列包含基因 ID。
library(msigdbr)
msig_t2g <- msigdbr(species = "Mus musculus", category = "H", subcategory = NULL)
msig_t2g <- msig_t2g[, colnames(msig_t2g) %in% c("gs_name", "entrez_gene")]
msig_t2g[1:3, ]

然后我们运行基因集富集,使用我们创建的术语到基因映射作为 enricher 函数中的 TERM2GENE 参数。
hallmark <- enricher(gene = genesWithPeakInTSS, universe = allGeneIDs, TERM2GENE = msig_t2g)
hallmark_df <- data.frame(hallmark)
hallmark_df[1:3, ]

我们在RNAseq课程中了解到了goseq包,这是另一个类似于clusterProfiler的功能标注包,在这里,我们对 MSigDB Hallmark 基因集执行相同的富集测试。
对于 goseq,我们需要所有基因(宇宙)的命名向量,其中 1 或 0 代表基因是否在 TSS 中达到峰值。我们可以简单地使用 as.integer 函数将逻辑向量转换为 1 表示 TRUE 和 0 表示 FALSE。
allGenesForGOseq <- as.integer(allGeneIDs %in% genesWithPeakInTSS)
names(allGenesForGOseq) <- allGeneIDs
allGenesForGOseq[1:3]
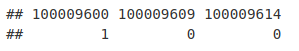
首先,我们必须使用 nullp 函数构建一个 nullp data.frame 以便在 goseq 中使用,并提供我们的命名向量、要使用的基因组和使用的基因标识符。
nullp 函数试图纠正我们在基因集测试中可能看到的基因长度偏差。也就是说,较长的基因可能有更多机会在其中出现峰值。
library(goseq)
pwf = nullp(allGenesForGOseq, "mm10", "knownGene", plot.fit = FALSE)

我们可以使用与用于 clusterProfiler 的基因映射相同的术语(尽管它必须从 tibble 转换为 goseq 的数据框)来运行基因集富集测试。
Myc_hallMarks <- goseq(pwf, "mm10", "knownGene", gene2cat = data.frame(msig_t2g))
Myc_hallMarks[1:3, ]









 苏公网安备32011502012024号
苏公网安备32011502012024号