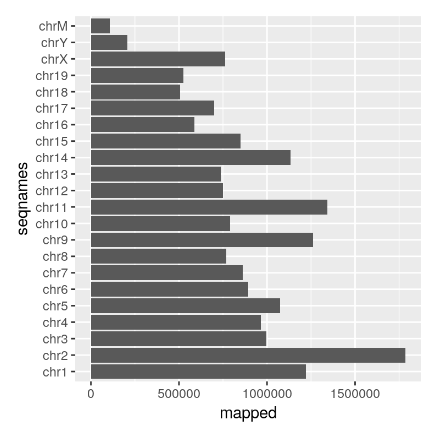
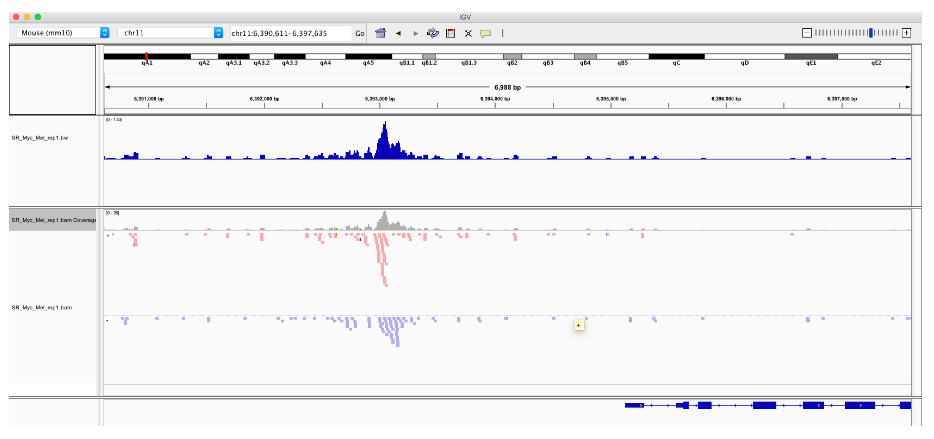
![]() 表观组
251 人阅读
|
0 人回复
|
2024-07-04
表观组
251 人阅读
|
0 人回复
|
2024-07-04
数据科学工厂 阅读 337 0 赞
数据科学工厂 阅读 297 0 赞
数据科学工厂 阅读 257 0 赞
数据科学工厂 阅读 226 0 赞
数据科学工厂 阅读 253 0 赞
数据科学工厂 阅读 232 0 赞
数据科学工厂 阅读 269 0 赞
数据科学工厂 阅读 297 0 赞
数据科学工厂 阅读 266 0 赞
数据科学工厂 阅读 280 0 赞



数据科学工厂 2024-11-28
数据科学工厂 2024-11-27
数据科学工厂 2024-11-26
数据科学工厂 2024-11-25
数据科学工厂 2024-11-24
数据科学工厂 2024-11-23
数据科学工厂 2024-11-20
数据科学工厂 2024-11-18
数据科学工厂 2024-11-15
数据科学工厂 2024-11-15





 苏公网安备32011502012024号
苏公网安备32011502012024号