动动发财的小手,点个赞吧!
1. ChIPseq 简介
染色质免疫沉淀,然后进行深度测序 (ChIPseq) 是一种成熟的技术,可以在全基因组范围内识别转录因子结合位点和表观遗传标记。
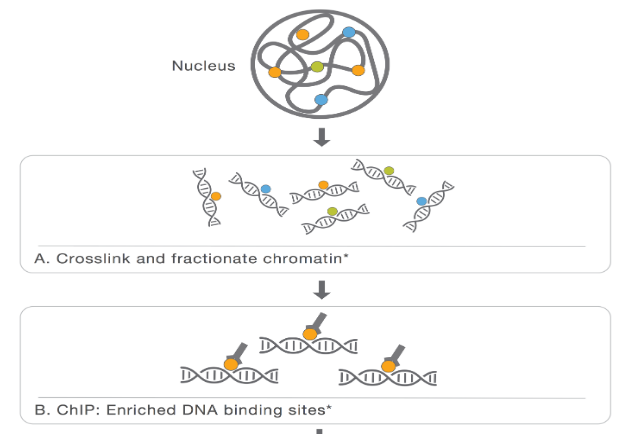
1.1. 实验处理
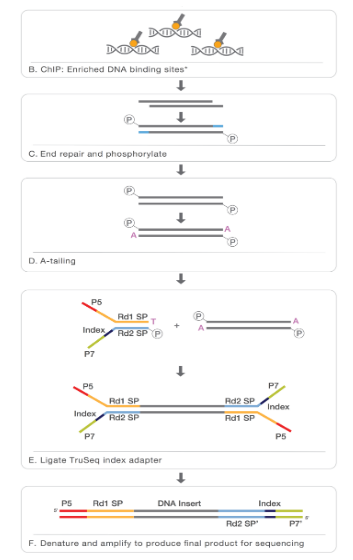
- 交联和蛋白质结合的 DNA。
- 通过抗体富集特定蛋白质或 DNA 。
- 添加 末端修复、A 尾和 Illumina adapters。
- 从任一端/两端测序。
2. 数据格式
原始 ChIPseq 测序数据将采用 FASTQ 格式。

在此 ChIPseq 研讨中,我们将研究小鼠 MEL 和 Ch12 细胞系中转录因子 Myc 的全基因组结合模式。
我们可以从 Encode 网站检索原始测序数据。在这里,我们使用小鼠 MEL 细胞系、样品 ENCSR000EUA(重复 1)下载 Myc ChIPseq 的测序数据。
3. 数据处理
3.1. 处理准备
一旦我们下载了原始 FASTQ 数据,我们就可以使用 ShortRead 包来检查我们的序列数据质量。
首先我们加载 ShortRead 库。
library(ShortRead)
我们将使用 ShortRead 包中的函数查看原始测序读数。这类似于我们为 RNAseq 执行的 QC。
不需要查看文件中的所有 reads 即可了解数据质量。我们可以简单地查看 reads 的子样本并节省一些时间和内存。
请注意,当我们进行子采样时,我们会从整个 FASTQ 文件中检索随机 reads。这很重要,因为 FASTQ 文件通常按其在测序仪上的位置排序。
3.2. 数据读取
我们可以使用 ShortRead 包中的函数从 FASTQ 文件中进行子采样。
在这里,使用 FastqSampler 和 yield 函数从 FASTQ 文件中随机抽取定义数量的 reads。在这里,我们对 100 万次 reads 进行了子采样。这应该足以了解数据的质量。
fqSample <- FastqSampler("~/Downloads/ENCFF001NQP.fastq.gz", n = 10^6)
fastq <- yield(fqSample)
生成的对象是一个 ShortReadQ 对象,显示有关循环数、reads 中的碱基对和内存中的 reads 数的信息。
fastq

3.3. 数据质控
如果愿意,我们可以使用我们熟悉的访问器函数来评估 FASTQ 文件中的信息。
- sread() - 检索 reads 序列。
- quality() - 检索 reads 质量作为 ASCII 分数。
- id() - 检索 reads 的 ID。
readSequences <- sread(fastq)
readQuality <- quality(fastq)
readIDs <- id(fastq)
readSequences

3.4. 质量检查
我们可以为我们的子采样 FASTQ 数据检查一些简单的质量指标。首先,我们可以查看整体读取的质量分数。
我们将 alphabetScore() 函数与我们的读取质量一起使用,以检索子样本中每个读取的总和质量。
readQuality <- quality(fastq)
readQualities <- alphabetScore(readQuality)
readQualities[1:10]

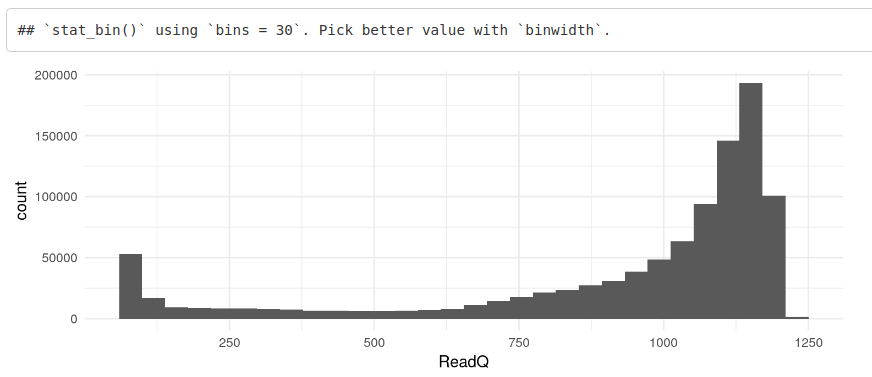
然后我们可以生成质量分数的直方图,以更好地了解分数的分布。
library(ggplot2)
toPlot <- data.frame(ReadQ = readQualities)
ggplot(toPlot, aes(x = ReadQ)) + geom_histogram() + theme_minimal()
3.5. 碱集频率
我们可以分别使用 alphabetFrequency() 和 alphabetByCycle() 函数查看 reads 中 DNA 碱基的出现以及整个测序周期中 DNA 碱基的出现。在这里,我们检查序列读取中 A、G、C、T 和 N(未知碱基)的总体频率。
readSequences <- sread(fastq)
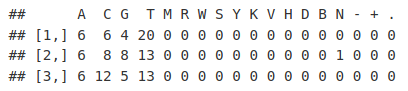
readSequences_AlpFreq <- alphabetFrequency(readSequences)
readSequences_AlpFreq[1:3, ]
一旦我们在序列读取中获得了 DNA 碱基的频率,我们就可以检索所有读取的总和。
summed__AlpFreq <- colSums(readSequences_AlpFreq)
summed__AlpFreq[c("A", "C", "G", "T", "N")]

3.6. 数据评估
我们可以使用 alphabetByCycle() 函数按循环查看 DNA 碱基出现情况。
readSequences_AlpbyCycle <- alphabetByCycle(readSequences)
readSequences_AlpbyCycle[1:4, 1:10]
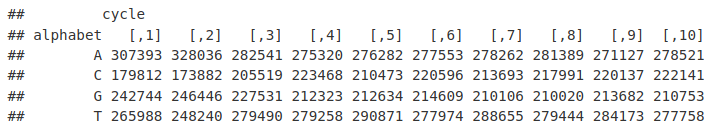
我们经常绘制此图以可视化循环中的碱基发生情况,以观察任何偏差。首先我们将基频排列成一个数据框。
AFreq <- readSequences_AlpbyCycle["A", ]
CFreq <- readSequences_AlpbyCycle["C", ]
GFreq <- readSequences_AlpbyCycle["G", ]
TFreq <- readSequences_AlpbyCycle["T", ]
toPlot <- data.frame(Count = c(AFreq, CFreq, GFreq, TFreq), Cycle = rep(1:36, 4),
Base = rep(c("A", "C", "G", "T"), each = 36))
现在我们可以使用 ggplot2 绘制频率
ggplot(toPlot, aes(y = Count, x = Cycle, colour = Base)) + geom_line() + theme_bw()
我们还可以评估周期内的平均读取质量。这将使我们能够确定是否存在质量随时间下降的问题。
为此,我们首先使用 as(read_quality,“matrix”) 函数将我们的 ASCII 质量分数转换为数字质量分数。
qualAsMatrix <- as(readQuality, "matrix")
qualAsMatrix[1:2, ]

我们现在可以使用箱线图可视化跨周期的质量。
boxplot(qualAsMatrix[1:1000, ])
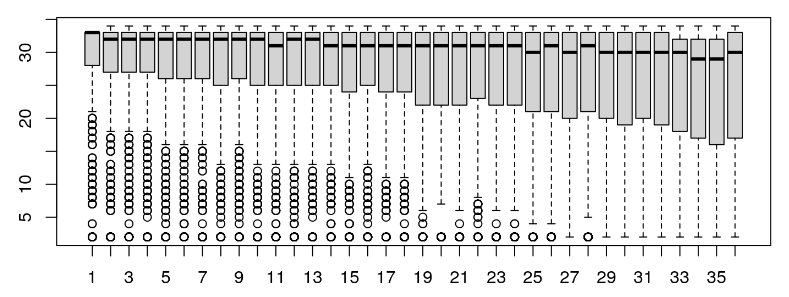
在这种情况下,reads 质量分数和 read 质量随时间的分布看起来还不错。我们经常希望一起访问 FASTQ 样本,以查看是否有任何样本符合这些指标。在这里,我们观察到第二批低质量分数,因此将删除一些质量分数低和未知碱基高的读数。
4. 数据过滤
我们将希望节省内存使用量,以允许我们处理加载大文件。这里我们设置了一个 FastqStreamer 对象来一次读入 100000 次读取。
fqStreamer <- FastqStreamer("~/Downloads/ENCFF001NQP.fastq.gz", n = 1e+05)
现在我们遍历文件,过滤读取并写出过滤读取的 FASTQ。我们正在过滤低质量的读数和具有许多非特异性 (N) 碱基调用的读数。
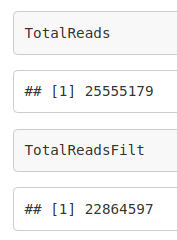
TotalReads <- 0
TotalReadsFilt <- 0
while (length(fq <- yield(fqStreamer)) > 0) {
TotalReads <- TotalReads + length(fq)
filt1 <- fq[alphabetScore(fq) > 300]
filt2 <- filt1[alphabetFrequency(sread(filt1))[, "N"] < 10]
TotalReadsFilt <- TotalReadsFilt + length(filt2)
writeFastq(filt2, "filtered_ENCFF001NQP.fastq.gz", mode = "a")
}








 苏公网安备32011502012024号
苏公网安备32011502012024号