学习目标
- 了解生成比较结果所需的步骤(Wald 检验)
- 总结不同层次的基因过滤
- 了解对数倍变化收缩
结果探索
默认情况下,DESeq2 使用 Wald 检验来识别在两个样本之间差异表达的基因。给定设计公式中使用的因素,以及存在多少个因素水平,我们可以为许多不同的比较提取结果。在这里,我们将介绍如何从 dds 对象获取结果,并提供一些有关如何解释它们的解释。
注意:Wald 检验也可用于连续变量。如果设计公式中提供的感兴趣变量是连续值,则报告的 log2FoldChange 是该变量的每单位变化。
1. 指定比较
在我们的数据集中,我们有三个样本类别,因此我们可以进行三种可能的成对比较:
- 对照 vs. Mov10 过表达
- 对照 vs. Mov10 敲除
- Mov10 过表达 vs. Mov10 敲除
我们只对上面的1 和2 感兴趣。当我们最初创建我们的 dds 对象时,我们提供了 ~ sampletype 作为我们的设计公式,表明 sampletype 是我们感兴趣的主要因素。
为了表明我们有兴趣比较哪两个样本,我们需要指定对比。用 DESeq2 results() 函数的输入以提取所需的结果。
对比可以用两种不同的方式指定(第一种方法更常用):
- 对比可以作为具有三个元素的字符向量提供:设计公式中(感兴趣的)因素的名称,要比较的两个因素水平的名称。最后给出的因子水平是比较的基准水平。语法如下:
contrast <- c("condition", "level_to_compare", "base_level")
results(dds, contrast = contrast)
- 对比可以作为 2 个字符向量的列表给出:折叠的名称随兴趣级别的变化而变化,折叠的名称随基本级别的变化而变化。这些名称应该与
resultsNames(object) 的元素完全匹配。
resultsNames(dds)
contrast <- list(resultsNames(dds)[1], resultsNames(dds)[2])
results(dds, contrast = contrast)
或者,如果你只有两个因子水平,你什么也做不了,也不必担心指定对比(即结果(dds))。在这种情况下,DESeq2 将根据水平的字母顺序选择您的基本因子水平。
首先,我们要评估 MOV10 过表达样本和对照样本之间的表达变化。因此,我们将使用第一种方法来指定对比并创建一个字符向量:
contrast_oe <- c("sampletype", "MOV10_overexpression", "control")
2. 结果
现在我们已经创建了对比,我们可以将其用作 results() 函数的输入。
res_tableOE <- results(dds, contrast=contrast_oe, alpha = 0.05)
注意:对于我们的分析,除了对比参数之外,我们还将为 alpha 参数提供 0.05 的值。当我们谈论基因级过滤时,我们将更详细地描述这一点。
返回给我们的结果是一个 DESeqResults 对象,它是 DataFrame 的一个简单子类。在许多方面,它可以像数据框一样对待(即在访问/子集数据时),但是重要的是要认识到下游步骤(如可视化)存在差异。
现在让我们看看结果中存储了哪些信息:
res_tableOE %>%
data.frame() %>%
View()

我们可以使用 mcols() 函数来提取有关存储在每列中的值代表什么的信息:
mcols(res_tableOE, use.names=T)
baseMean: 所有样本的归一化计数的平均值log2FoldChange: log2倍变化lfcSE: 标准误差stat: Wald 统计pvalue: Wald 检验 p-valuepadj: BH adjusted p-values
3. P-values
p 值是用于确定是否有证据拒绝原假设的概率值。较小的 p 值意味着有更强有力的证据支持备择假设。然而,因为我们正在对每个单独的基因进行测试,所以我们需要更正这些 p 值以进行多次测试。
结果中的 padj 列代表针对多重检验调整的 p 值,是结果中最重要的一列。通常,padj < 0.05 等阈值是识别重要基因的良好起点。 DESeq2 中多重测试校正的默认方法是 Benjamini-Hochberg 错误发现率 (FDR) 的实现。还有其他可用的校正方法,可以通过将 pAdjustMethod 参数添加到 results() 函数来更改。
4. Filter
仔细看看我们的结果。当我们浏览它时,您会注意到对于选定的基因,pvalue和 padj 列中有 NA 值。这是什么意思?

缺失值表示已作为 DESeq() 函数的一部分进行过滤的基因。在进行差异表达分析之前,忽略那些很少或根本没有机会被检测为差异表达的基因是有益的。这将增加检测差异表达基因的能力。 DESeq2不会从原始计数矩阵中删除任何基因,因此所有基因都将出现在您的结果表中。 DESeq2 遗漏的基因满足以下三个过滤标准之一:
- 所有样本中计数为零的基因
如果在一行中,所有样本的计数均为零,则没有表达信息,因此不会测试这些基因。
res_tableOE[which(res_tableOE$baseMean == 0),] %>%
data.frame() %>%
View()
这些基因的 baseMean 列将为零,log2 倍数变化估计、p 值和调整后的 p 值都将设置为 NA。
- 具有极端计数异常值的基因
DESeq() 函数为每个基因和每个样本计算异常值的诊断测试,称为库克距离。 Cook 距离衡量单个样本对基因的拟合系数的影响程度,Cook 距离的较大值旨在指示异常值计数。包含库克距离高于阈值的基因被标记,但是标记至少需要 3 个重复,因为很难判断哪个样本可能是异常值,只有 2 个重复。我们可以通过在 results() 函数中使用 cooksCutoff 参数来关闭此过滤。
res_tableOE[which(is.na(res_tableOE$pvalue) &
is.na(res_tableOE$padj) &
res_tableOE$baseMean > 0),] %>%
data.frame() %>%
View()
- 具有低平均归一化计数的基因
DESeq2 定义了一个低均值阈值,它是根据您的数据凭经验确定的,其中重要基因的比例可以通过减少考虑进行多重测试的基因数量来增加。这是基于这样一种观念,即计数非常低的基因通常由于高度分散而不太可能看到显著差异。
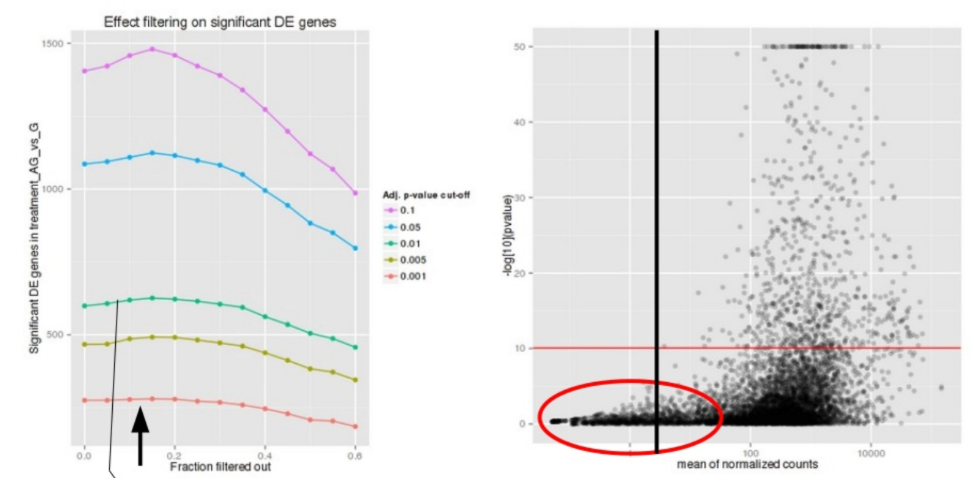
在用户指定的值 (alpha = 0.05),DESeq2 评估显著基因数量的变化,因为它根据基因的平均计数过滤掉越来越大的基因部分,如上图所示。娴熟基因数量达到峰值的点是用于过滤经过多次测试的基因的低平均阈值。还有一个参数是通过设置 independentFiltering = F 来关闭过滤。
res_tableOE[which(!is.na(res_tableOE$pvalue) &
is.na(res_tableOE$padj) &
res_tableOE$baseMean > 0),] %>%
data.frame() %>%
View()
注意:默认情况下,DESeq2 将执行上述过滤;但是其他 DE 工具(例如 EdgeR)不会。过滤是必要的步骤,即使您使用的是 limma-voom 和/或 edgeR 的拟似然法。在使用其他工具时,请务必遵循预过滤步骤,如 Bioconductor 上的用户指南中所述,因为它们通常表现得更好。
5. Fold change
结果中的另一个重要列是 log2FoldChange。对于大量的基因列表,很难提取有意义的生物学相关性。为了帮助提高严格性,还可以添加倍数变化阈值。请记住,在设置该值时,我们正在处理 log2 倍数变化,因此 log2FoldChange < 1 的截止值将转化为实际倍数变化 2。
结果中的倍数变化计算如下:
log2 (normalized_counts_group1 / normalized_counts_group2)
问题是,这些倍数变化估计并不完全准确,因为它们没有考虑到我们在低读取计数下观察到的离散。为了解决这个问题,需要调整 log2 倍的变化。
LFC
为了生成更准确的 log2 foldchange (LFC) 估计值,DESeq2 允许在基因信息较低时将 LFC 估计值收缩至零,这可能包括:
LFC 收缩使用来自所有基因的信息来生成更准确的估计。具体来说,所有基因的 LFC 估计值的分布(作为先验)用于将信息很少或高度分散的基因的 LFC 估计值缩小到更有可能(较低)的 LFC 估计值。
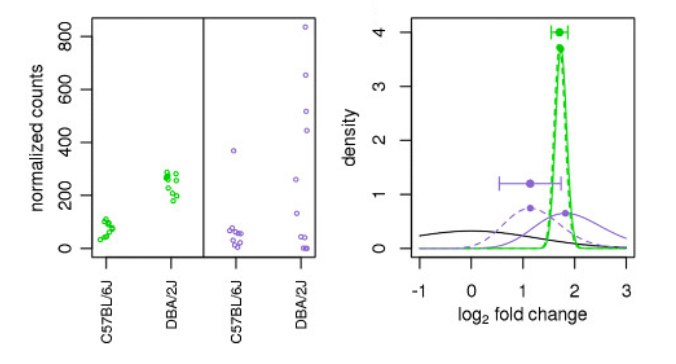
在上图中,我们有一个使用绿色基因和紫色基因的例子。对于每个基因,绘制了两种不同小鼠品系(C57BL/6J 和 DBA/2J)中每个样本的表达值。两个基因对于两个样本组具有相同的平均值,但绿色基因在组内几乎没有变异,而紫色基因具有高水平的变异。对于组内变异低的绿色基因,未收缩的 LFC 估计(绿色实线的顶点)与收缩的 LFC 估计(绿色虚线的顶点)非常相似。然而,由于高度分散,LFC 对紫色基因的估计有很大不同。因此,即使两个基因可以具有相似的归一化计数值,它们也可以具有不同程度的 LFC 收缩。请注意,LFC 估计值向先验值收缩(黑色实线)。
缩小 log2 倍变化不会改变被识别为显著差异表达的基因总数。倍数变化的收缩是为了帮助下游评估结果。例如,如果您想根据倍数变化对重要基因进行子集化以进行进一步评估,您可能需要使用收缩值。此外,对于需要折叠变化值作为输入的 GSEA 等功能分析工具,您可能希望提供收缩值。
要生成缩小的 log2 倍变化估计值,您必须使用函数 lfcShrink() 在您的结果对象(我们将在下面创建)上运行一个额外的步骤。
res_tableOE_unshrunken <- res_tableOE
# Apply fold change shrinkage
res_tableOE <- lfcShrink(dds, coef="sampletype_MOV10_overexpression_vs_control", type="apeglm")
根据您使用的 DESeq2 版本,收缩估计的默认方法会有所不同。如上所述,可以通过在 lfcShrink() 函数中添加参数类型来更改默认值。对于大多数最新版本的 DESeq2,type="normal" 是默认值,并且是早期版本中的唯一方法。已经表明,在大多数情况下,存在比“正常”方法偏差更小的替代方法,因此我们选择使用 apeglm。
MA plot
可用于探索我们的结果的图是 MA 图。 MA 图显示了归一化计数的平均值与所有测试基因的 log2 倍数变化的关系。显著 DE 的基因被着色以便于识别。这也是说明 LFC 收缩效果的好方法。 DESeq2 包提供了一个简单的函数来生成 MA 图。
让我们从未收缩的结果开始:
plotMA(res_tableOE_unshrunken, ylim=c(-2,2))
收缩的结果
plotMA(res_tableOE, ylim=c(-2,2))
在左侧,您绘制了未收缩的倍数变化值,您可以看到低表达基因的大量分散。也就是说,许多低表达者表现出非常高的倍数变化。收缩后,我们看到倍数变化估计要小得多。
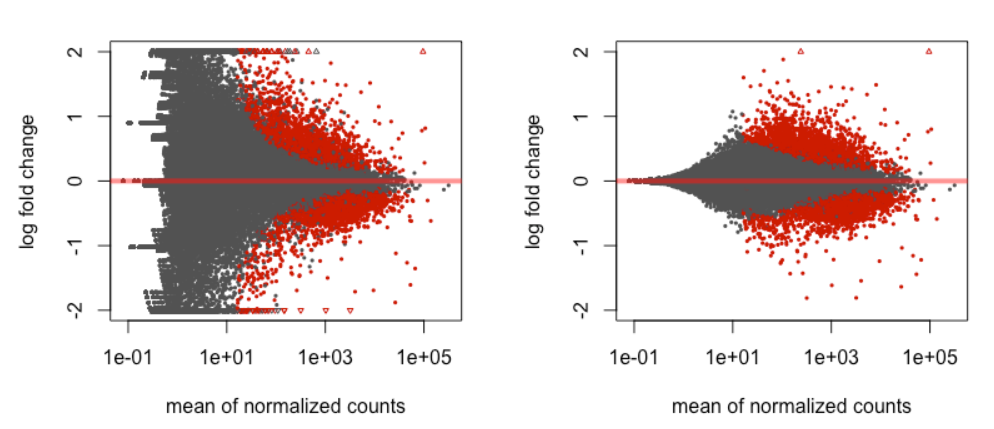
除了上述比较之外,该图还允许我们评估倍数变化的幅度以及它们相对于平均表达的分布方式。通常,我们希望在整个表达水平范围内看到重要的基因。








 苏公网安备32011502012024号
苏公网安备32011502012024号