
本期教程

一步构建模式植物OrgDb数据库
source("../Set_OrgDb_Database.R")
# 使用函数
Set_OrgDb_Database(
emapper_file = "out.emapper_tomato.csv", ## 输入的eggnog结果文件
json_file = "ko00001.json", ## 下载ko00001.json,下载网址:https://www.genome.jp/kegg-bin/get_htext?ko00001
tax_id = "4081", ## 物种信息
genus = "Solanum",
species = "lycopersicum",
versions = "4.0", ## 版本号
maintainer = "du<***@qq.com>", ## 修改为你的名字和邮箱
author = "du<***@qq.com>", ## 修改为你的名字和邮箱
outputDir = "."## 保存路径
)
获得本期教程文本文档,在后台回复:**20240802。注意:一步构建模式植物OrgDb数据库Set_OrgDb_Database()函数教程可在社群中获得**。请大家看清楚回复关键词,每天都有很多人回复错误关键词,我这边没时间和精力一一回复。
写在前面
前期,我们发表过模式植物GO背景基因集制作,这些教程也算是比较“经济实惠”、且常用的。在我们制作分析是会遇到的问题,随着前面教程推出,也有同学咨询如何制作KEGG背景文件。今天,自己也查了相关的教程,进行整理一下。最开始的目标是,将我们注释文件进行本地打包建库,使用时就会更加的方便。但是,在此过程中,出现一些问题,目前还未解决。那就安排到下一期的内容吧。
一、使用ggNOG-mapper仅需注释
1 模式植物KEGG注释
本次使用 <span class="ne-text">Egg-mapper</span>进行注释,网页版可以支持 <span class="ne-text">proteins</span>/<span class="ne-text">CDS</span>/<span class="ne-text">genomic</span>/<span class="ne-text">Metagenomic</span>和 <span class="ne-text">seeds</span>文件格式。
1.1 下载模式植物序列
本次事例中,我们使用 <span class="ne-text">Proteins</span>文件进行注释。使用番茄(tomato)蛋白序列,你可以使用其它序列。
wget https://solgenomics.net/ftp/genomes/Solanum_lycopersicum/annotation/ITAG4.0_release/ITAG4.0_proteins.fasta
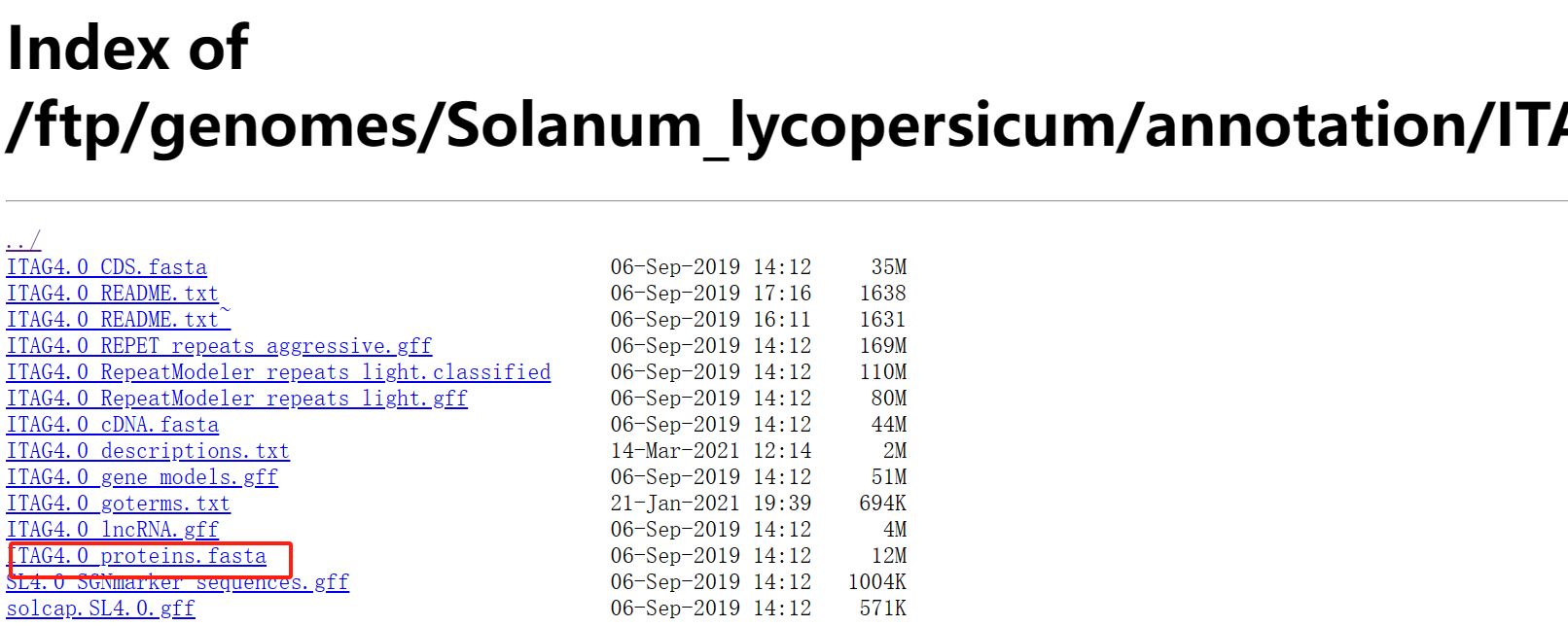
1.2 上传到 <strong><span class="ne-text">ggNOG-mapper</span></strong>
<span class="ne-text">egg-mapper</span>网址
http://eggnog-mapper.embl.de/

百度搜索
- 上传蛋白序列
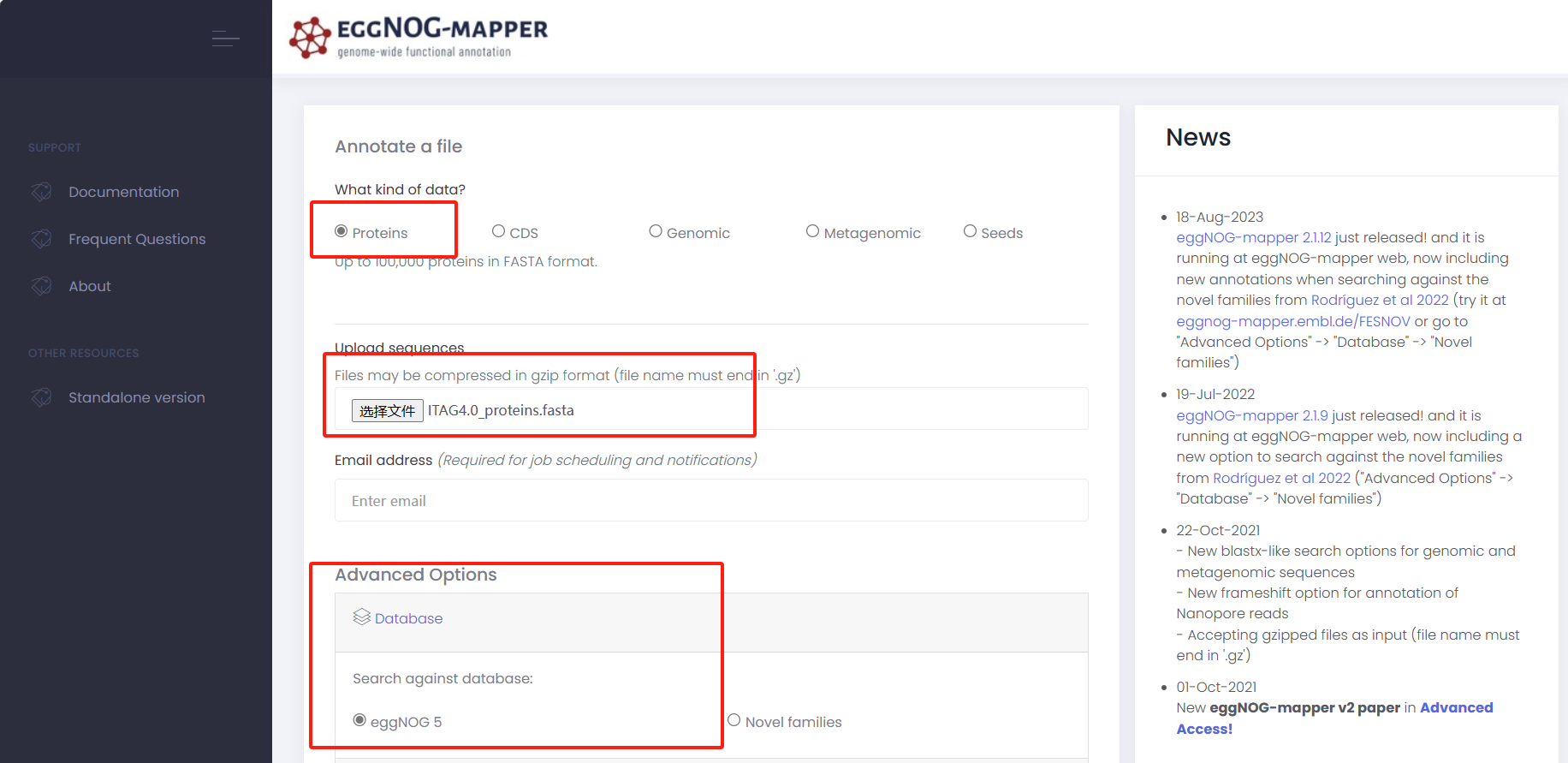
- 输入邮箱
- 提交
- 邮箱中收到eggNOG-mapper发送的邮件
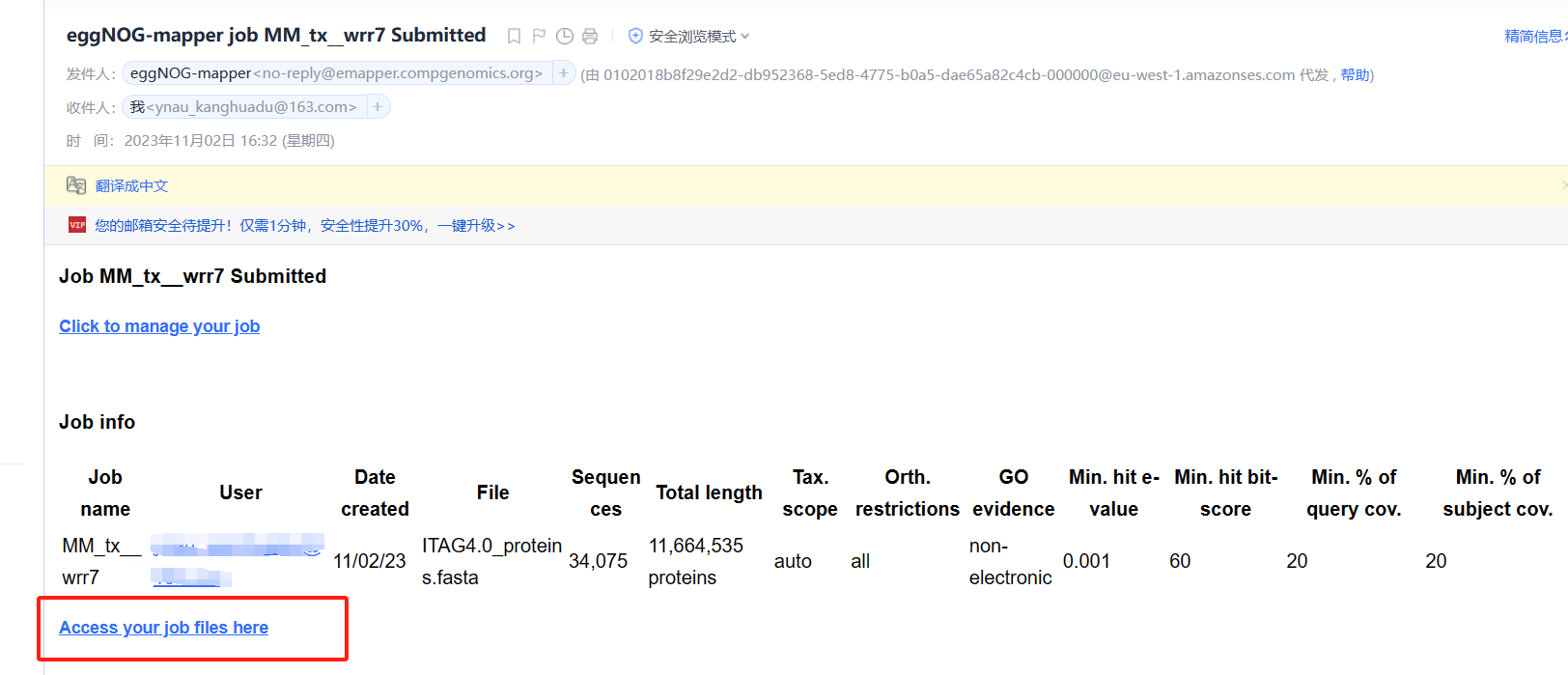
- 开始运行【重点】 在我们收到邮件时,很多同学可能会认为此任务已经开始(PS:自己也是这么认为的)。但是,我们需要点击
<span class="ne-text">Click to manage your job</span>进去工作台,点击 <span class="ne-text">start job</span>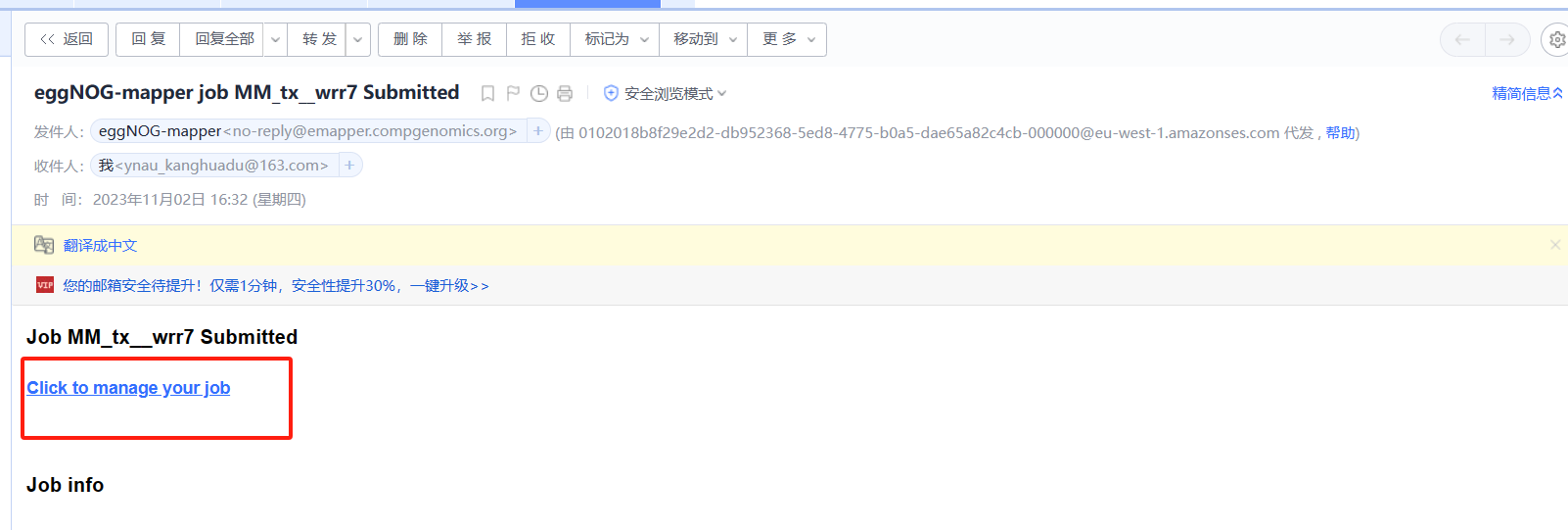 进入工作台
进入工作台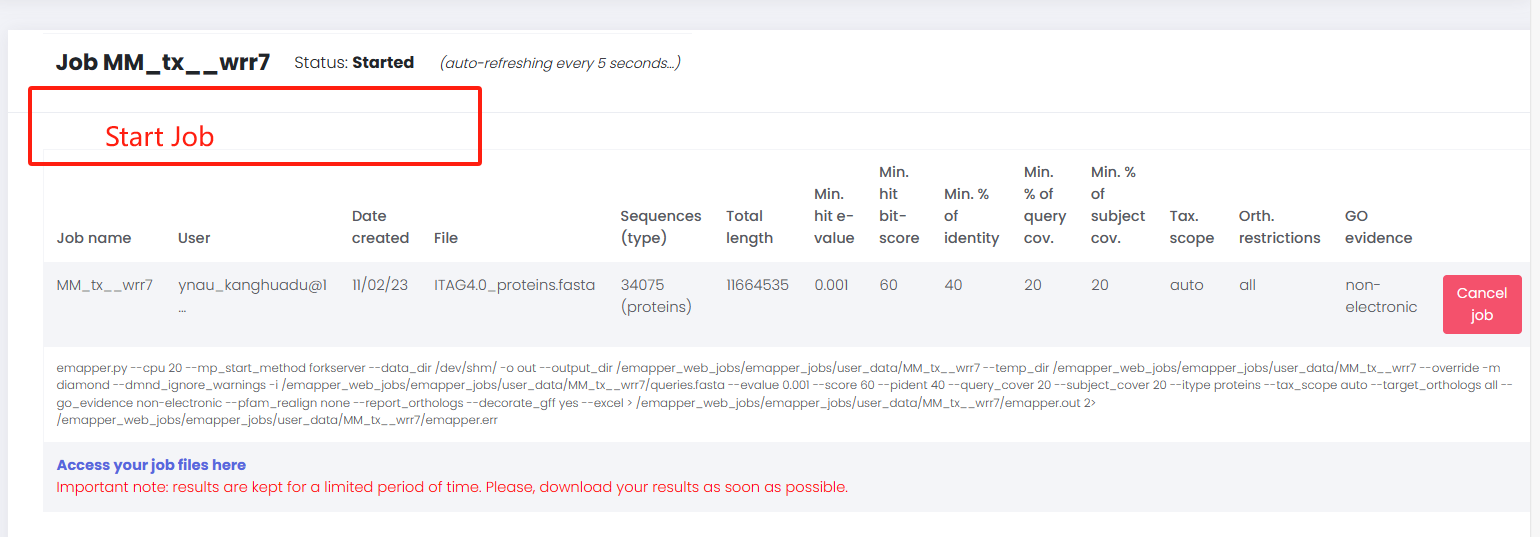 开始运行工作文件后台界面
开始运行工作文件后台界面
1.3 下载注释文件
若你提交的任务结束,可以到工作任务后台下载数据。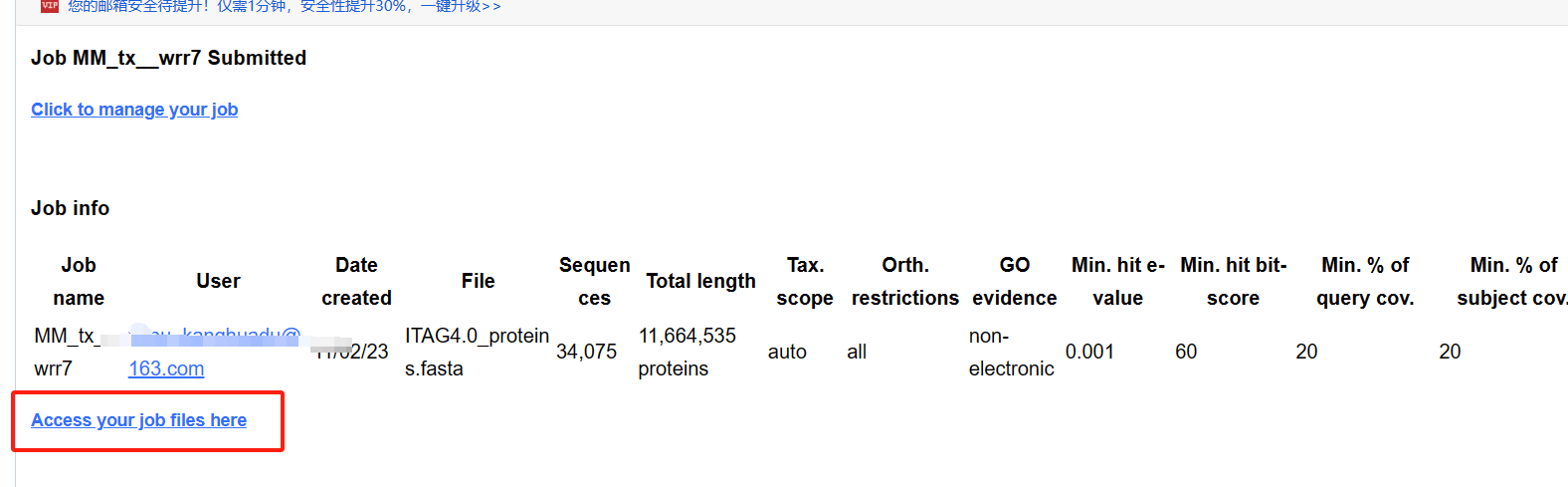 下载数据
下载数据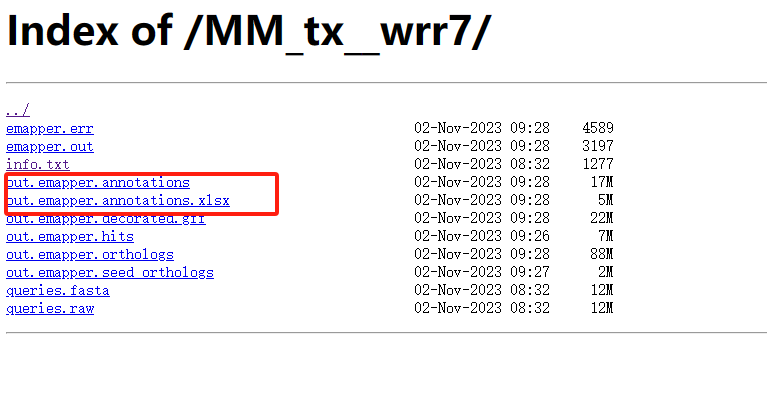
2 本地运行
2.1安装 <strong><span class="ne-text">egg-mapper</span></strong>软件
- conda或mamba安装
mamba install -c bioconda -c conda-forge eggnog-mapper
or
conda install -c bioconda -c conda-forge eggnog-mapper
- 源码安装
下载网址:
https://github.com/eggnogdb/eggnog-mapper/releases/tag/2.1.12
 3. 帮助文档网址
3. 帮助文档网址
https://github.com/eggnogdb/eggnog-mapper/wiki/
## v2.1.5版本帮助文档
https://github.com/eggnogdb/eggnog-mapper/wiki/eggNOG-mapper-v2.1.5-to-v2.1.12#user-content-Software_Requirements
2.2 安装软件需求
- Python 3.7 (or greater)
- BioPython 1.76 (python package) (BioPython 1.78 should work since emapper version 2.1.7)
- psutil 5.7.0 (python package)
- xlsxwriter 1.4.3 (python package), only if using the --excel option
- wget (linux command, required for downloading the eggNOG-mapper databases with download_eggnog_data.py, and to create new Diamond/MMseqs2 databases with create_dbs.py)
- sqlite (>=3.8.2)
2.3 硬盘储存需求
- eggNOG annotation databases:~40 GB
- eggNOG sequences:~9 GB
- MMseqs2 database of eggNOG sequences:~11 GB(~86 GB if MMseqs2 index is created)
- PFAM database:~3 GB
2.4 内存需求
- Using --dbmem loads the whole eggnog.db sqlite3 annotation database during the annotation step, and therefore requires ~44 GB of memory.
- Using the --num_servers option when running HMMER in server mode (a.k.a. hmmgpmd, which is used for -m hmmer --usemem, --pfam_realign denovo or hmm_server.py) loads the HMM database as many times as specified in the argument (e.g. --pfam_realign denovo --num_servers 2 loads the PFAM database into memory twice, with up to roughly 2 GB per instance).
2.5 数据库下载
下载网址:http://eggnog6.embl.de/#/app/downloads 在eggnog_6.0版本中提供此下载数据信息
在eggnog_6.0版本中提供此下载数据信息 使用软件自带的脚本下载数据库:
使用软件自带的脚本下载数据库:
download_eggnog_data.py -h
注意,我们这里使用 <span class="ne-text">conda</span>或 <span class="ne-text">mamba</span>,以及添加到\$PATH路径中,可以直接运行。若你使用源码安装,及未添加路径,需求添加 <span class="ne-text">python /PATH/download_eggnog_data.py -h</span>运行。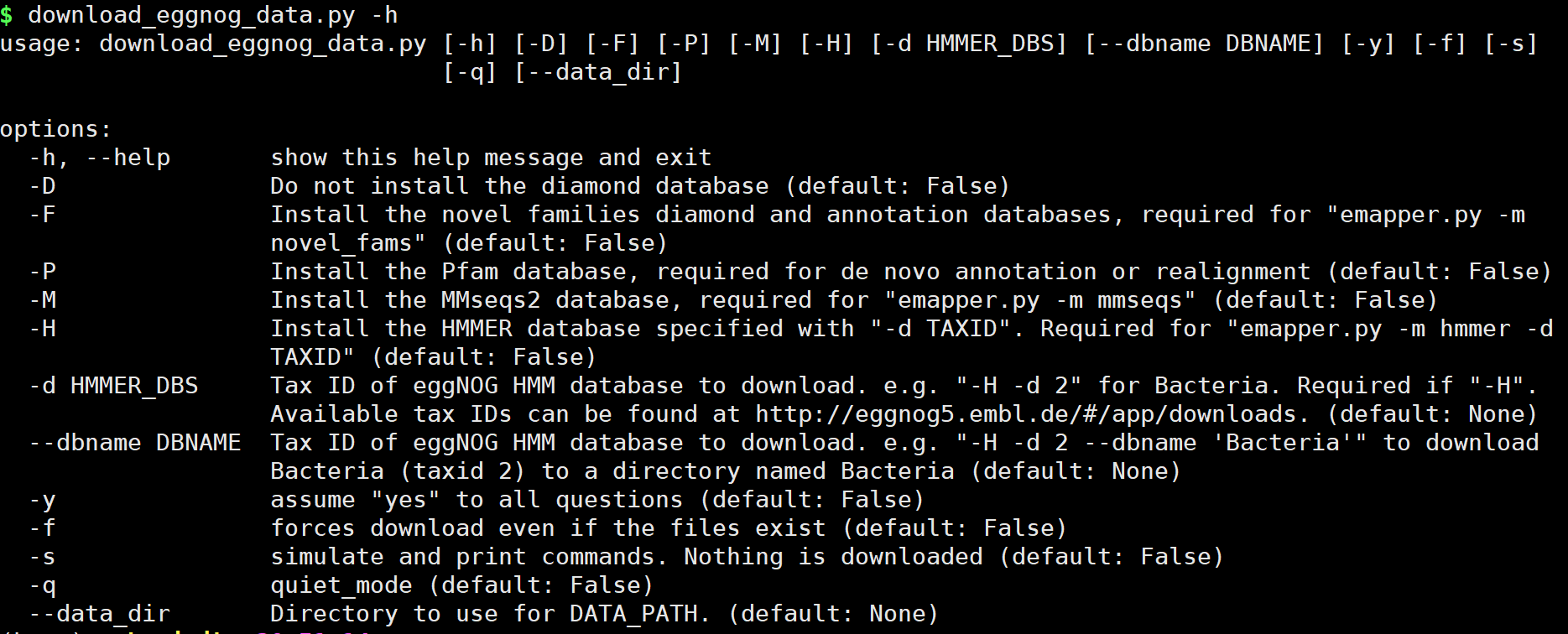 下载数据库整体命令:
下载数据库整体命令:
download_eggnog_data.py --data_dir /home/Data/DataBase/Eggno_Database/ -y -F -D -H
参数选择:
options:
-h, --help show this help message and exit
-D Do not install the diamond database (default: False)
-F Install the novel families diamond and annotation databases, required for "emapper.py -m novel_fams" (default: False)
-P Install the Pfam database, required for de novo annotation or realignment (default: False)
-M Install the MMseqs2 database, required for "emapper.py -m mmseqs" (default: False)
-H Install the HMMER database specified with "-d TAXID". Required for "emapper.py -m hmmer -d TAXID" (default: False)
-d HMMER_DBS Tax ID of eggNOG HMM database to download. e.g. "-H -d 2" for Bacteria. Required if "-H". Available tax IDs can be
found at http://eggnog5.embl.de/#/app/downloads. (default: None)
--dbname DBNAME Tax ID of eggNOG HMM database to download. e.g. "-H -d 2 --dbname 'Bacteria'" to download Bacteria (taxid 2) to a
directory named Bacteria (default: None)
-y assume "yes" to all questions (default: False)
-f forces download even if the files exist (default: False)
-s simulate and print commands. Nothing is downloaded (default: False)
-q quiet_mode (default: False)
--data_dir Directory to use for DATA_PATH. (default: None)
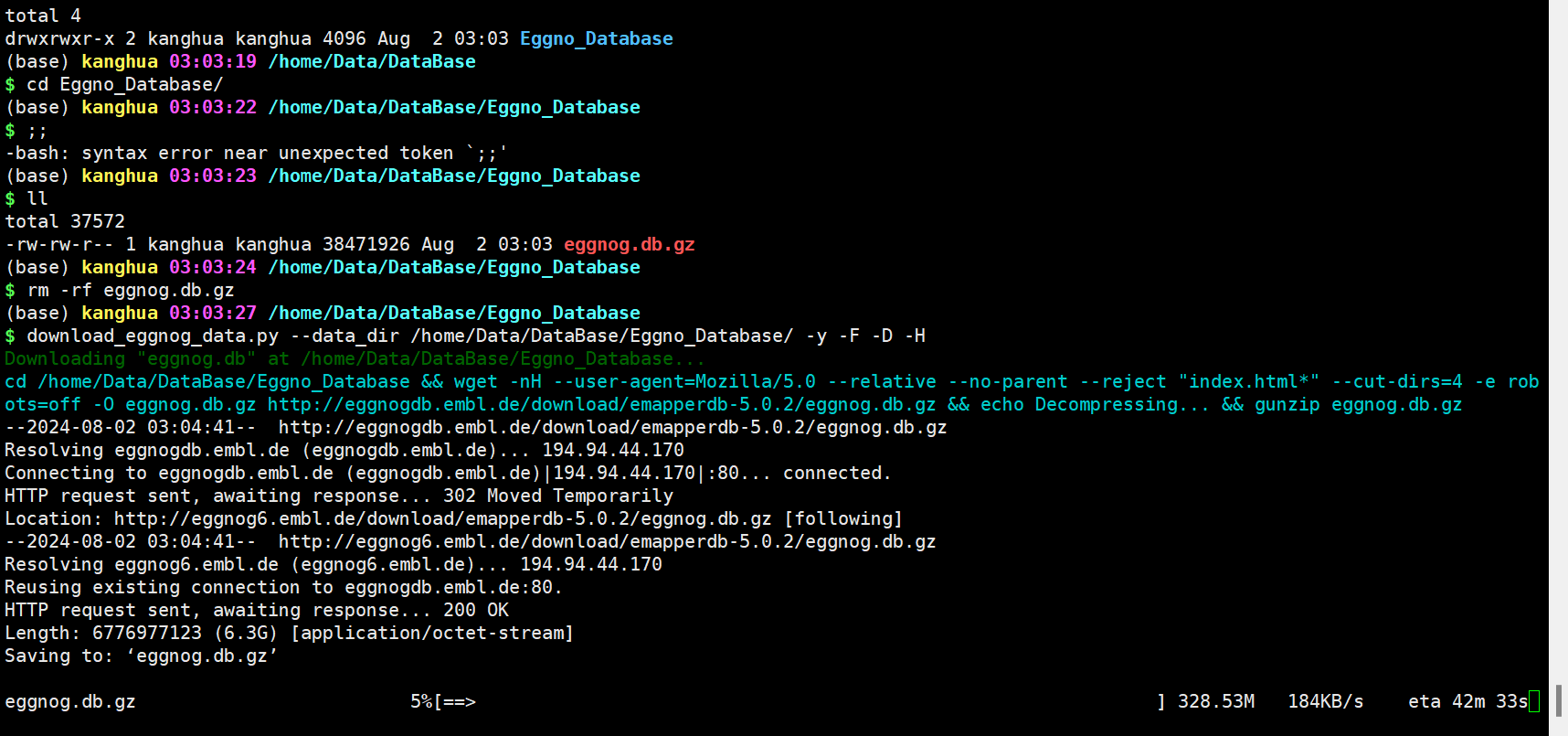
2.6 运行
HMMER方法本地检索细菌数据库 -i输入、—output输出文件前缀、-d指定数据库数据、—data_dir指定数据库位置
python emapper.py -i test/polb.fa --output polb_bact -d bact --data_dir ~/data/db/eggnog
diamond方法<span class="ne-text">-m</span>指定diamond方法,默认为hmmer方法。diamond在多于千条序列时才会体现速度优势,少量序列会感觉非常慢,而且结果也没有hmmer的更准确,尤其是对远源注释方面。
python emapper.py -i test/polb.fa --output diamond_bact_ -d bact --data_dir ~/data/db/eggnog -m diamond
此外,作者也在推文或帮助文档中说明,可以增加内存和线程数量,使用 <span class="ne-text">--usemem</span>增加内存,使用 <span class="ne-text">--cup</span>增加线程数,<span class="ne-text">--override</span>是强制覆盖结果。
python emapper.py -i test/polb.fa --output diamond_bact_ -d bact --data_dir ~/data/db/eggnog --usemem --cpu 10 --override -m diamond
跟多细节请查看官网即可:
https://github.com/eggnogdb/eggnog-mapper/wiki/eggNOG-mapper-v2.1.5-to-v2.1.12#user-content-Software_Requirements
对于使用在线 <strong><span class="ne-text">eggNOG-mapper</span></strong>还是自己在服务器中进行搭建呢?
对于这个问题,每个人分析的环境不同以及资源不同,我们根据自己的环境和资源进行合理调整即可。
- 若手中的有充足资源服务器,可以自己搭建,有利于后续流程的搭建和分析。
- 若自己手中并未有那么充足资源,其实使用在线的分析也可以,足够使用。
3 结果分析
运行成功后,我们获得一下注释文件信息。如何操作,就成为后续分析重点。类似思路,可以依据模式植物GO背景基因集制作。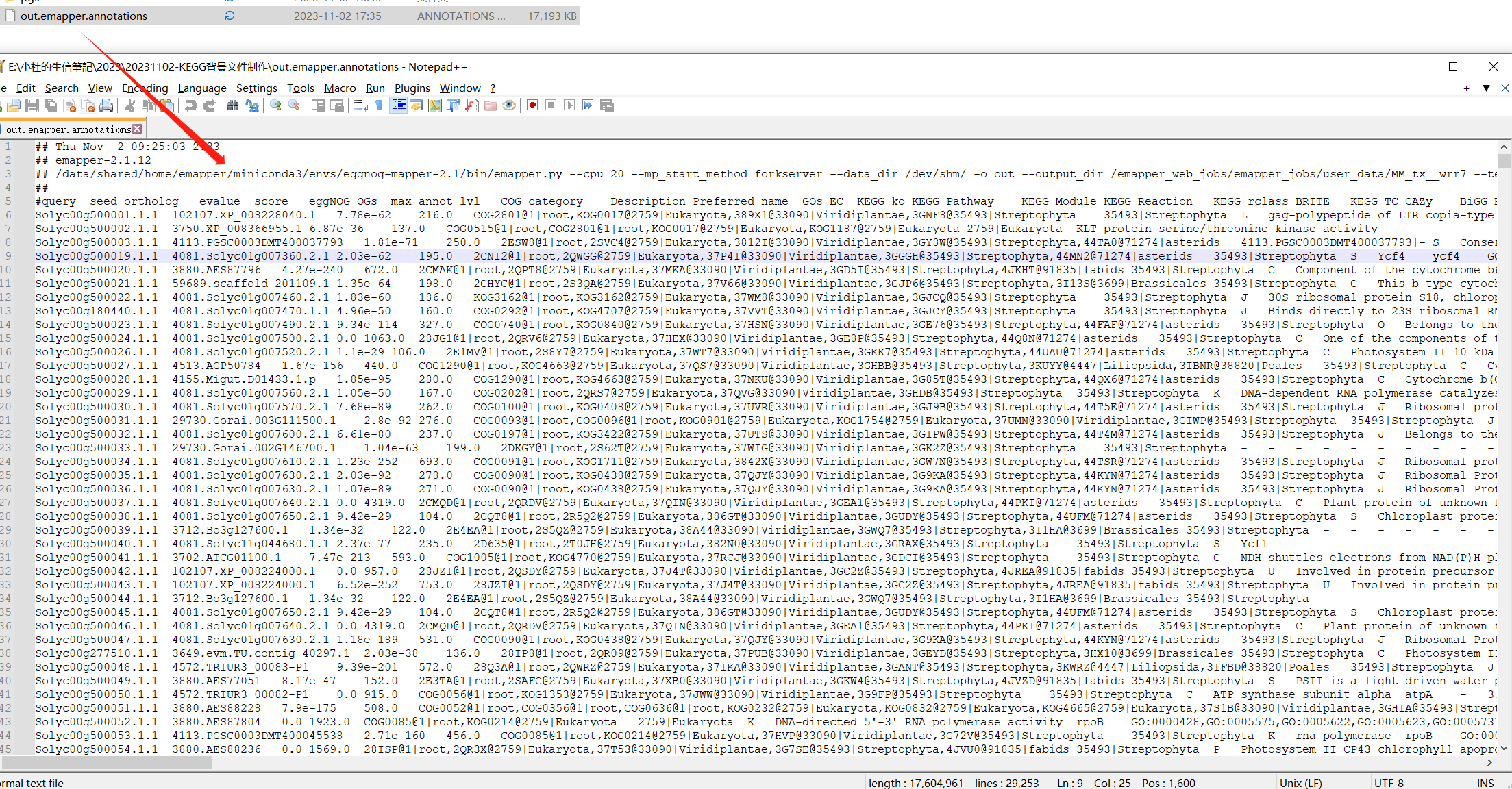 标注的信息就是我们制作KEGG背景文件重要的信息。
标注的信息就是我们制作KEGG背景文件重要的信息。
我们在这里可以发现,在egg-mapper中注视到 <span class="ne-text">GO ID</span>,那么是不是GO背景文件也可以使用这个文件进行制作呢??与前期的教程有何差异呢??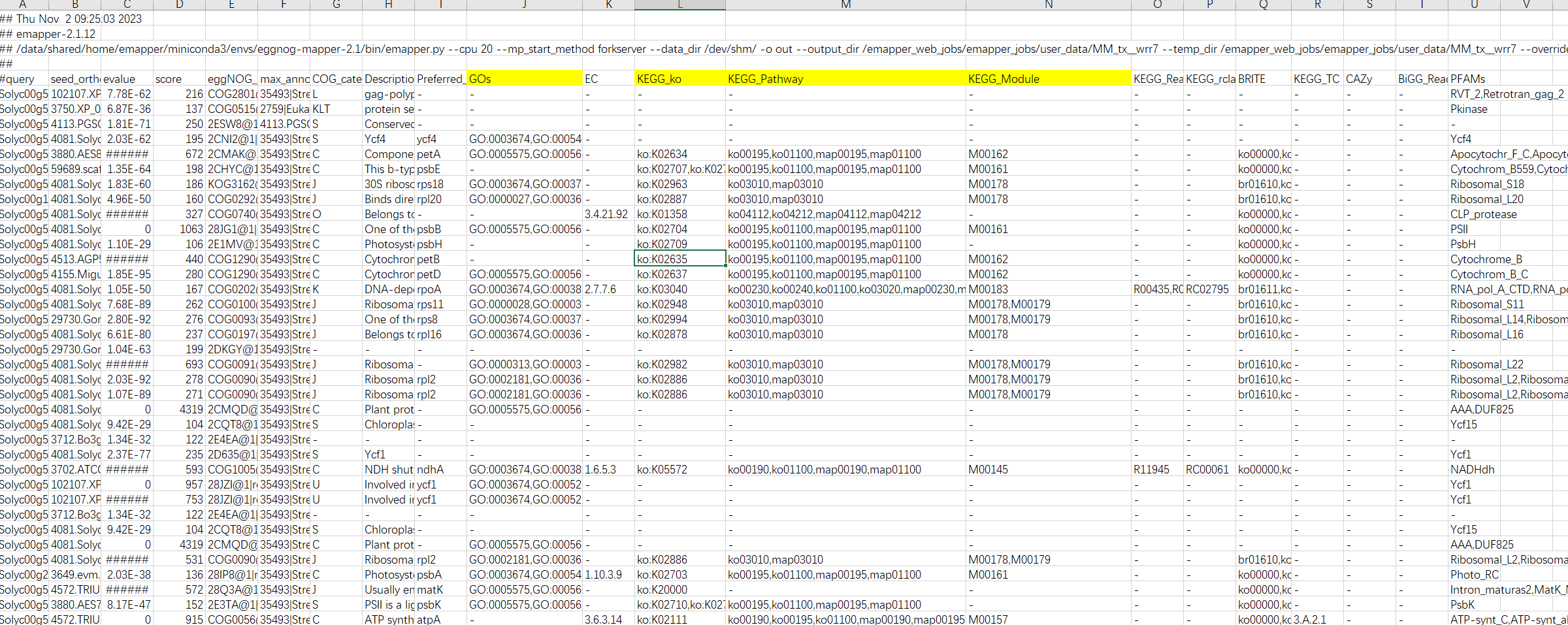 结果解读:
结果解读:
第一列:查询序列名称
第二列:eggNOG种子序列
第三列:eggNOG种子序列 evalue
第四列:eggNOG种子序列 bit score
第五列:匹配的OGs
第六列:最大的OG分类等级
第七列:COG功能分类
第八列:OGs最低分类等级物种描述
第九列:首选名称
第十列:GOs, 预测的GO,逗号分隔
第十一列:EC,预测额EC
第十二列:KEGG_KO: 预测的KO
第十三列:KEGG_Pathway:预测的ko通路
第十四列:KEGG_Module:预测的ko功能单元
第十五列:KEGG_Reaction:预测的酶促反应
第十六列:KEGG_rclass:预测的ko相关类
第十七列:BRITE:预测的brite功能分类
第十八列:KEGG_TC
第十九列:CAZy:预测的碳水化合物活性酶
第二十列:BiGG_Reaction:BiGG代谢反应预测
第二十一列:PFAMs:pfam蛋白预测
我们了解整个注释文件的信息,如何来提取对应的数据集分析呢?
3.1 使用TBtols进行富集
- 打开
<span class="ne-text">Tbtools</span>中的 <span class="ne-text">eggNOG-mapper</span>
- 将注释文件拖拽进去
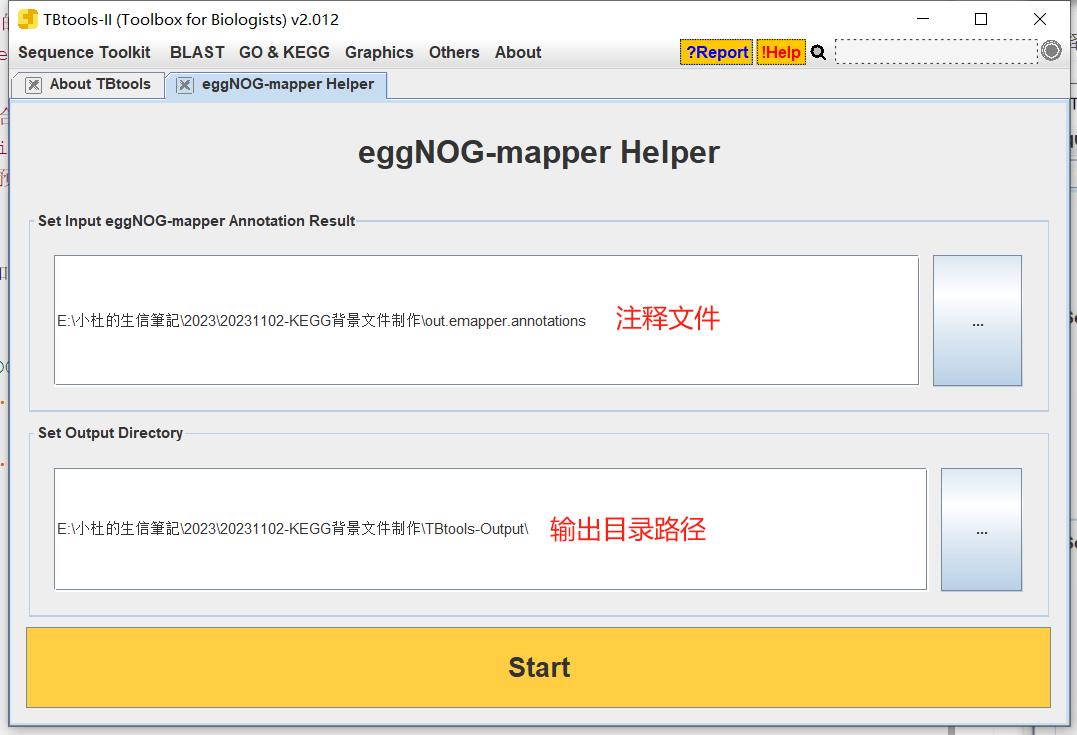
- 输出结果
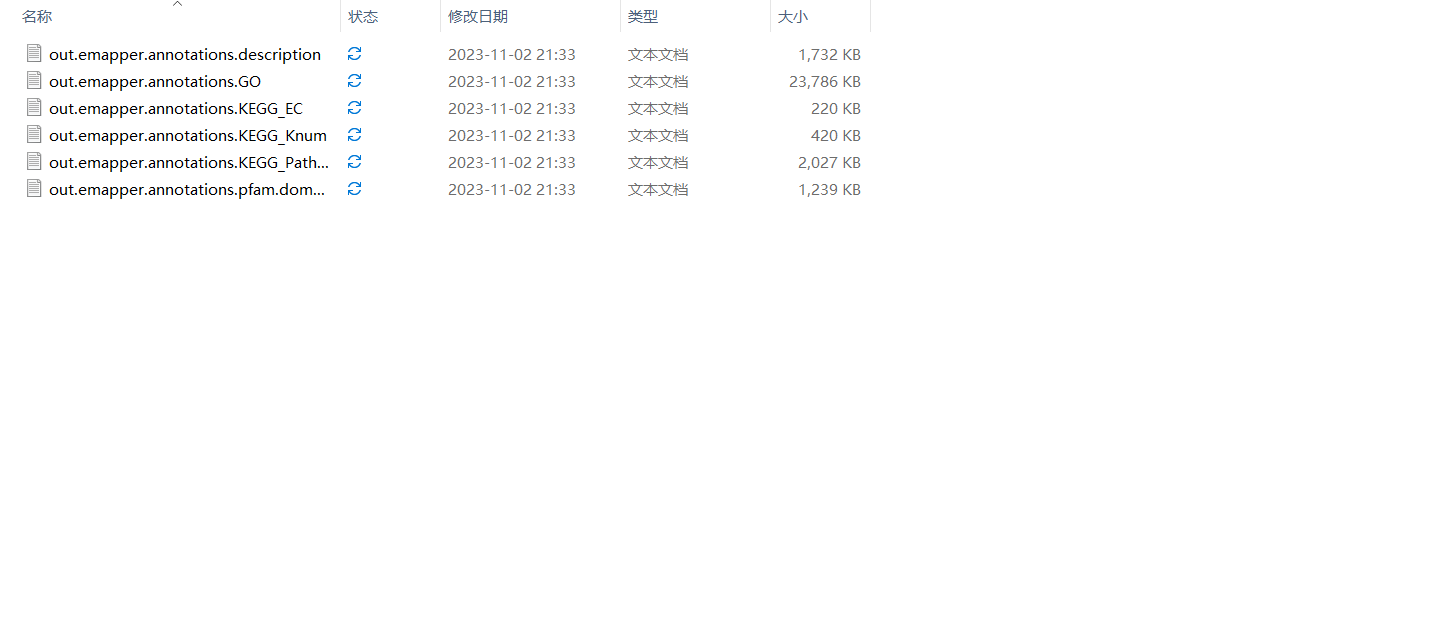 输出文件,将
输出文件,将 <span class="ne-text">注释ID</span>与每个的 <span class="ne-text">GO ID</span>或 <span class="ne-text">KEGG ID</span>进行整理,直接生成一个背景文件信息,可以直接上传到云平台中进行分析。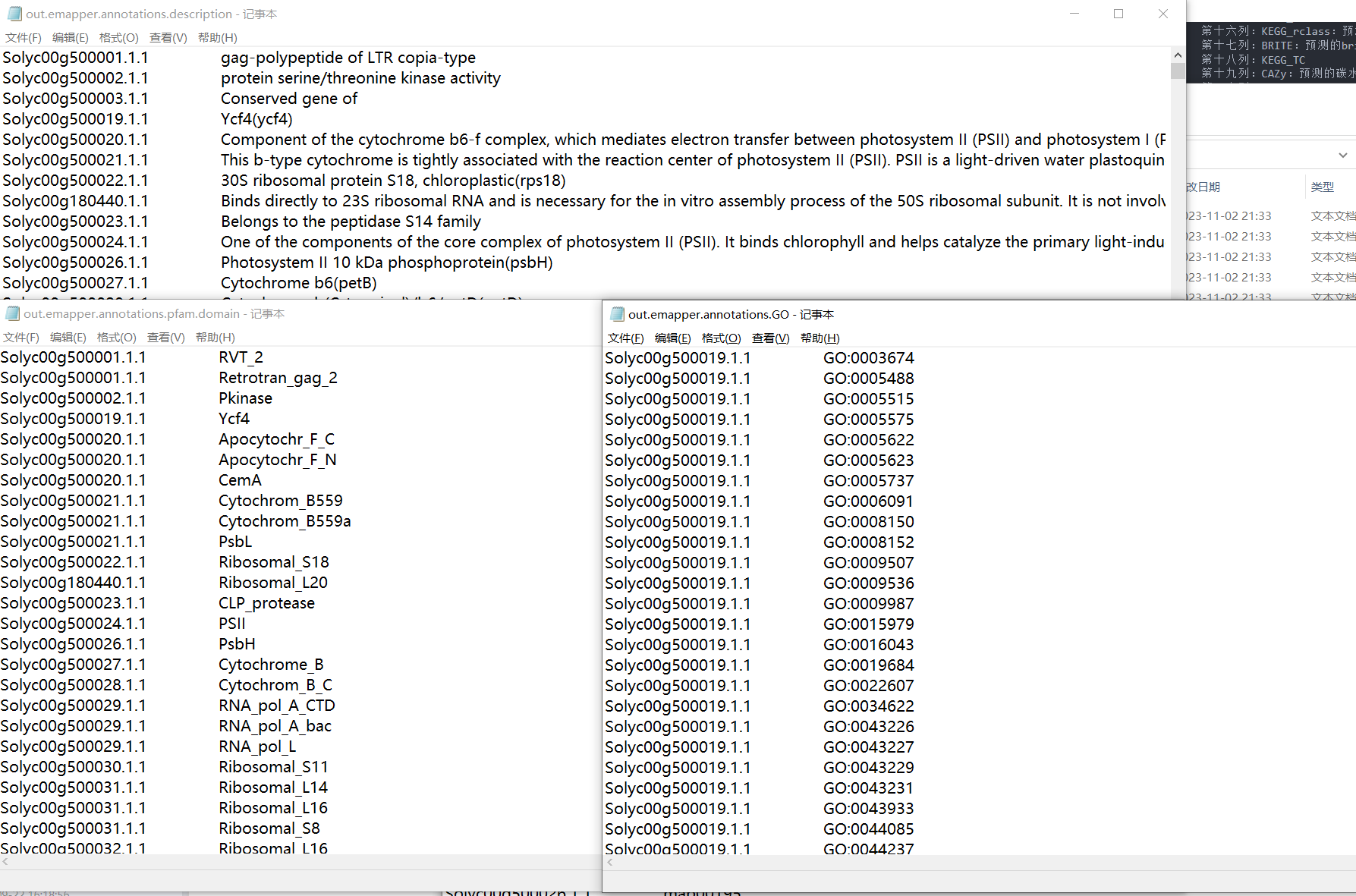 3.使用TBtools进行富集 TBtools为什么受这么多人的欢迎,功能很强大。基本把你遇到的问题,都会提供解决。 (1)下载
3.使用TBtools进行富集 TBtools为什么受这么多人的欢迎,功能很强大。基本把你遇到的问题,都会提供解决。 (1)下载 <span class="ne-text">.KEGG.BackEnd</span>文件和 <span class="ne-text">go-basic.obo</span>下载网址:
https://tbtools.cowtransfer.com/s/566e88227a0045
(2)运行 依次输入进行做好的注释文件和的差异基因即可进行富集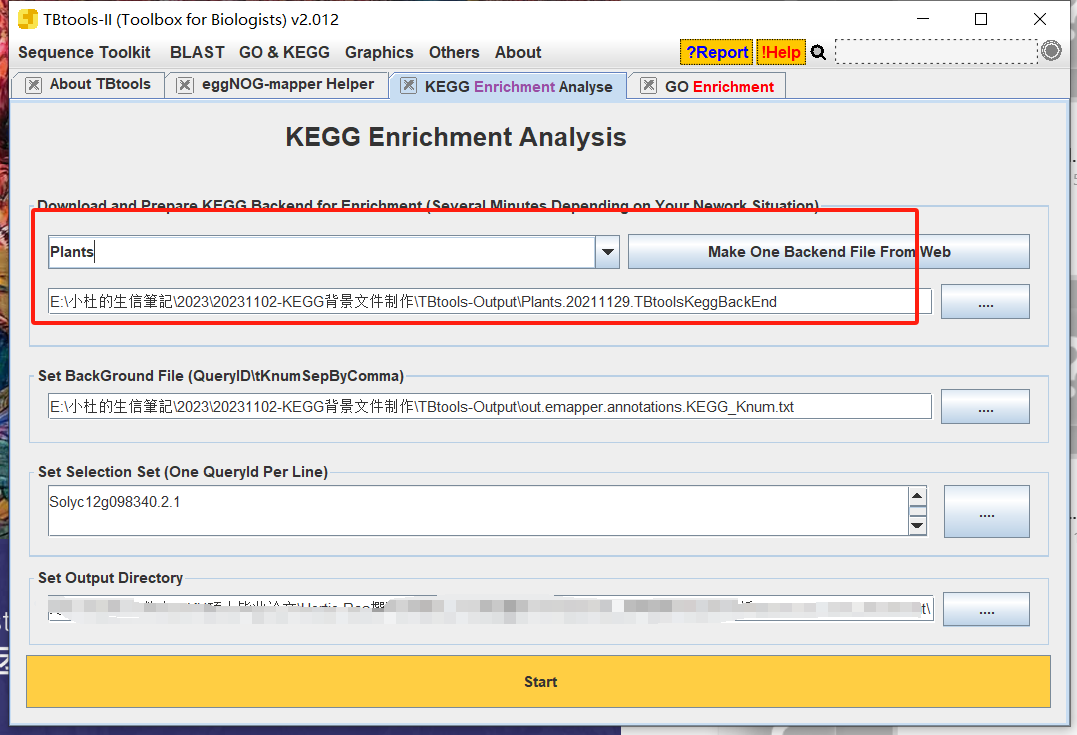
(3)结果 输出两个结果,一个是全部结果,另一个是过滤后的结果。
输出结果文件
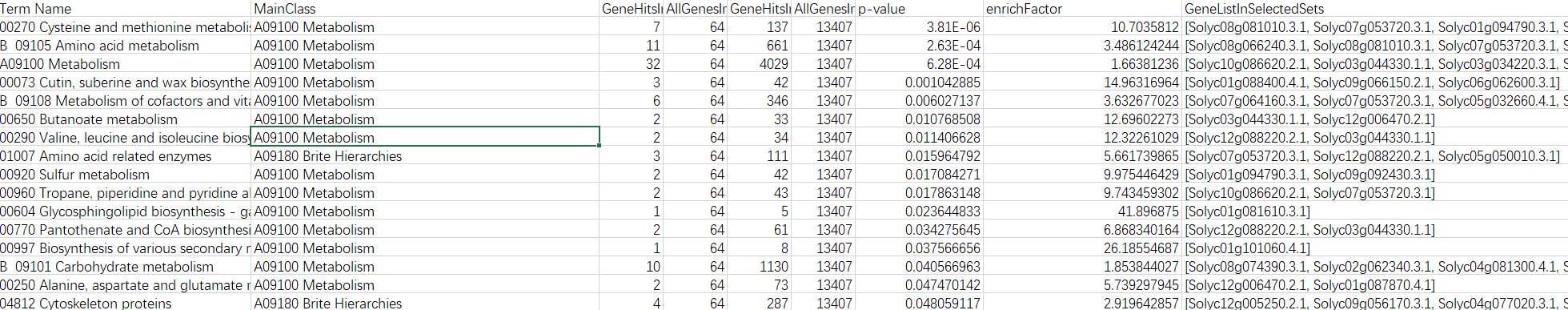
KEGG注释全部结果
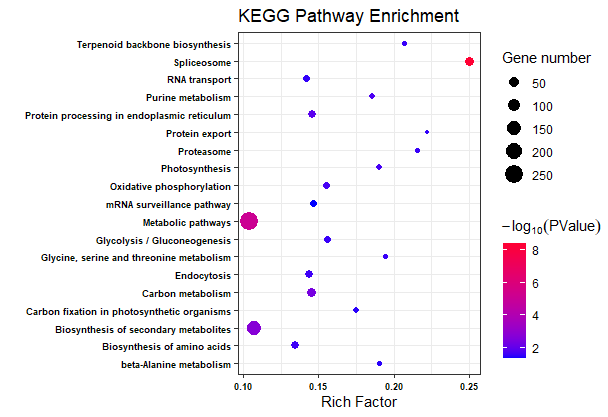
(4)绘制结果图形 绘制图形,可以根据自己的需求进绘制即可。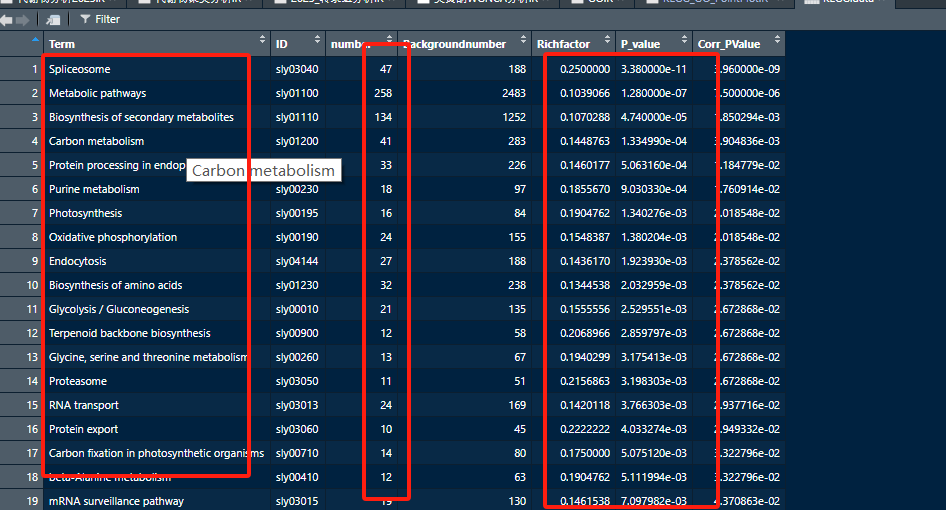 根据自己需求进行整理即可。
根据自己需求进行整理即可。
KEGG.data <- read.csv("KEGG_Enrichment_plotdata.csv", header = T)
names(KEGG.data) <- c("Term","ID","number", "Backgroundnumber","Richfactor","P_value", "Corr_PValue")
head(KEGG.data)
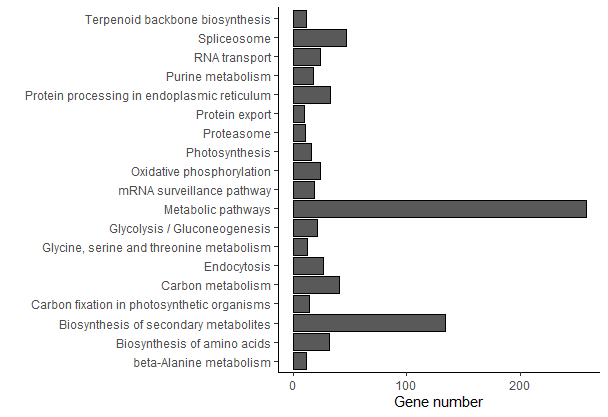
ggplot(KEGG.data, aes(x = number, y = Term))+
geom_bar(stat = "identity", position = "dodge", color = "black")+
xlab("Gene number")+ ylab("")+
theme_classic()+theme(legend.title = element_blank())
ggplot(KEGG.data, aes(x = Richfactor, y = Term))+
geom_point(aes(size = number, color = -1*log10(Corr_PValue)))+
scale_color_gradient(low = "#0000ff", high = "#ff0033")+
labs(color = expression(-log[10](PValue)), size = "Gene number",
x = "Rich Factor", y = "", title = "KEGG Pathway Enrichment")+
theme(legend.background = element_blank())+theme_bw()+ ## theme_bw() 去掉背景
theme(axis.text = element_text(face = "bold",color = "black", size = 7)+
## 修改字体大小,根据自己实际情况修改
theme(
axis.text = element_text(size = 12, family = "Arial", color = "black"),
axis.title = element_text(size = 14, family = "Arial", color = "black"),
legend.text = element_text(size = 12, family = "Arial", color = "black"),
legend.title = element_text(size = 14, family = "Arial", color = "black"))
)
参考:
- https://mp.weixin.qq.com/s/Ii-pB0JEDt\_pRRKPyq3RLw
- https://mp.weixin.qq.com/s/kIf6C2u3FID3ZeLtsB4eZQ
二、构建OrgDb数据库
我们上面的操作是将构建出来背景文件文件使用云平台或TBtools工具进行功能富集,那么若是使用 <span class="ne-text">clusterProfiler</span>等包进行本地富集,就需要构建OrgDb数据库。
我们这里基于网上的教程资源,整理 <span class="ne-text">如何制作模式植物本地OrgDB数据库</span>。在文末会标注的相关参考资源,在此也感谢各位知识分享博主的分享。
2.1 整理ggNOG-mapper注释文件
在注释文件中,会有很多列信息,我们可以做直接导入数据(PS:可能导入时会报错)。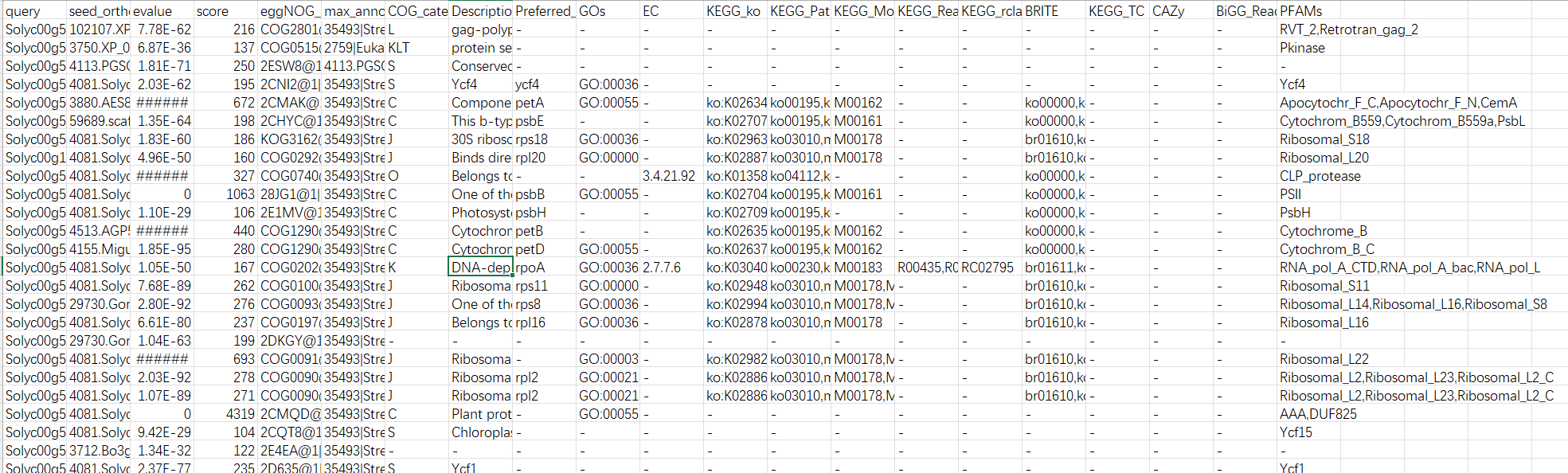
emapper <- read.table("out.emapper_tomato.txt", header = T, sep = "\t")
若是报错,我们可以在本地提取相关的信息即可。我们所需的信息就只有 <span class="ne-text">ID</span>,<span class="ne-text">GO</span>,<span class="ne-text">KEGG</span>信息列。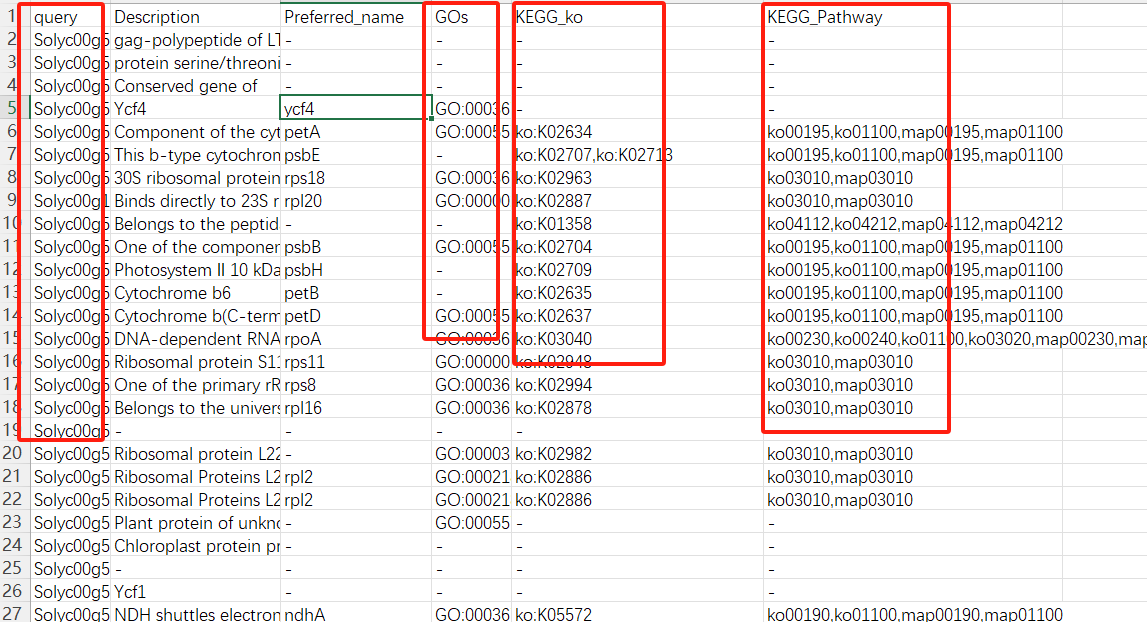
emapper <- read.csv("out.emapper_tomato.csv",header = T, check.names = F)
2.2 温馨提示
本教程,我们会提供分布式的步骤教程,大家可以直接跟随教程直接做即可。每个步骤,我们都会有详细的说明。此外,**我们也会提供 <strong><span class="ne-text">一步式教程</span></strong>,方便我们后期的制作 ,**<span class="ne-text">一步式教程</span>,在我们的社群中可以查看。
模式植物本地OrgDb数据库制作
- 导入相关的R包
library(clusterProfiler)
library(tidyverse)
library(stringr)
library(KEGGREST)
library(AnnotationForge)
library(clusterProfiler)
library(dplyr)
library(jsonlite)
library(purrr)
library(RCurl)
options(stringsAsFactors = F)
- 导入数据
setwd("D:\\小杜的生信笔记\\2024\\20240802_模式植物构建OegDB数据库")
emapper <- read.csv("out.emapper_tomato.csv",header = T, check.names = F)
head(emapper)
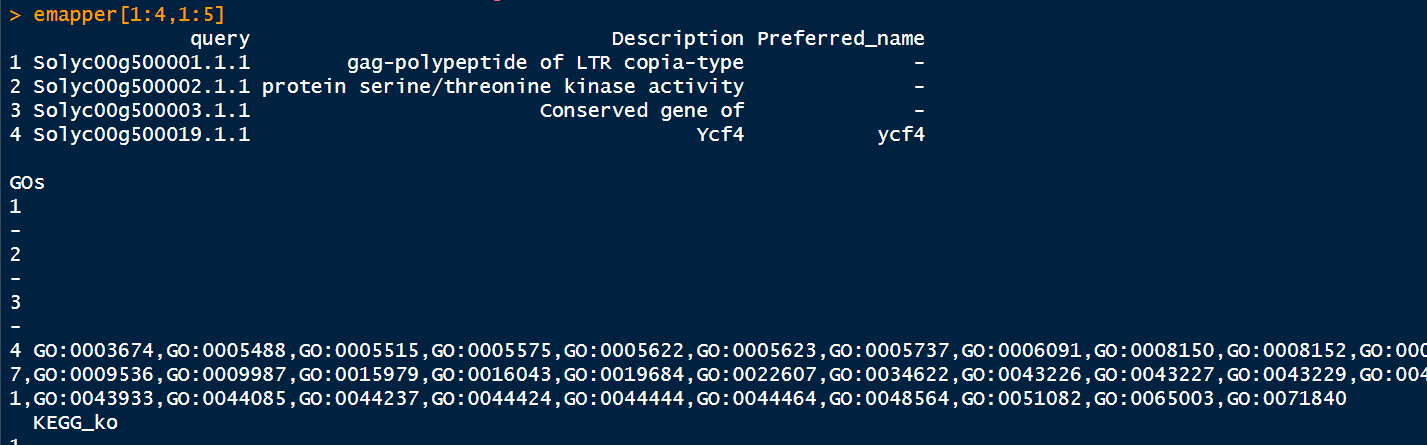 将缺省值替换为NA
将缺省值替换为NA
emapper[emapper==""]<-NA
- 提取GO信息
gene_info <- emapper %>%
dplyr::select(GID = query,GENENAME = Preferred_name) %>%
na.omit()
gos <- emapper %>%
dplyr::select(query, GOs) %>%
na.omit()
head(gos)
- 构建空的gene2go数据框
gene2go = data.frame(GID =character(),
GO = character(),
EVIDENCE = character())
排顺整理,**需要花费一定时间。**
for (row in1:nrow(gos)) {
the_gid <- gos[row, "query"][[1]]
the_gos <- str_split(gos[row,"GOs"], ",", simplify = FALSE)[[1]]
df_temp <- data_frame(GID=rep(the_gid, length(the_gos)),
GO = the_gos,
EVIDENCE = rep("IEA", length(the_gos)))
gene2go <- rbind(gene2go, df_temp)
}
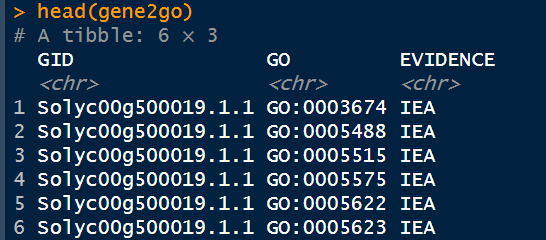 5. 删除NA行
5. 删除NA行
gene2go$GO[gene2go$GO=="-"]<-NA
gene2go<-na.omit(gene2go)
- 提取KEGG信息 在这里,我们提取的是
<span class="ne-text">KO号列</span>。
#把Emapper中的query列、KEGG_ko列提出出来
gene2ko <- emapper %>%
dplyr::select(GID = query,
Ko = KEGG_ko) %>%
na.omit()
#将gene2ko的Ko列中的“Ko:”删除,不然后面找pathway会报错
gene2ko$Ko <- gsub("ko:","",gene2ko$Ko)
head(gene2ko)
> head(gene2ko)
GID Ko
1 Solyc00g500001.1.1 -
2 Solyc00g500002.1.1 -
3 Solyc00g500003.1.1 -
4 Solyc00g500019.1.1 -
5 Solyc00g500020.1.1 K02634
6 Solyc00g500021.1.1 K02707,K02713
- 下载KO的json文件,下载地址https://www.genome.jp/kegg-bin/get_htext?ko00001
下载后,放在 <span class="ne-text">路劲文件夹</span>中。

由于文本限制,后面的教程请看教程二
...............................
参考资源:
- https://www.jianshu.com/p/693d66681710
- https://www.jianshu.com/p/bb4281e6604e
- https://www.jianshu.com/p/9c9e97167377
- https://bioconductor.org/packages/release/bioc/vignettes/clusterProfiler/inst/doc/clusterProfiler.html#supported-organisms
❝
若我们的教程对你有所帮助,请 <span class="ne-text">点赞+收藏+转发</span>,这是对我们最大的支持。
往期部分文章
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
2. 精美图形绘制教程
3. 转录组分析教程
4. 转录组下游分析
小杜的生信筆記** ,主要发表或收录生物信息学教程,以及基于R分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!**






 苏公网安备32011502012024号
苏公网安备32011502012024号