代码来源于论文
A complex phenotype in salmon controlled by a simple change in migratory timing
lastz和multiz可以通过conda进行安装
我用拟南芥的基因组做测试
运行脚本
bash ~/my_data/raw_data/practice/science.salmon/lastz_seed_and_chain.sh abc an1chr1 c24chr1 an1.chr1.fa c24.chr1.fa
第一个位置参数是输出文件夹
第二个位置参数是需要比对的基因组序列id (target)
第三个位置参数是需要比对的基因组序列id(query)
第四个位置参数是需要比对的基因组(target)
第五个位置参数是需要比对的基因组(query)
(2 3 位置参数序列id不能是一样的,如果两个序列id一样就会报错,我在这里搞了好长时间)
输出的内容
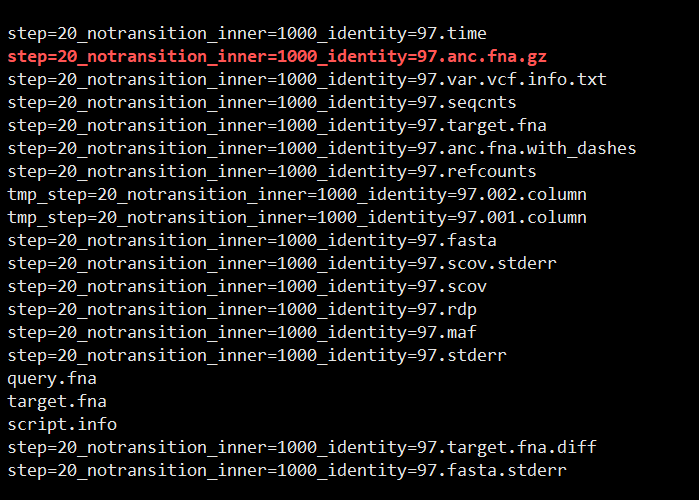
001.column是参考的结果
002.column是查询的结果
接下来是R语言的代码
source("extract_variants_between_two_seqs.R")
library(tidyverse)
tq_seqs<-tibble(ts=toupper(read_lines("abc/tmp_step=20_notransition_inner=1000_identity=97.001.column")),qs=toupper(read_lines("abc/tmp_step=20_notransition_inner=1
000_identity=97.002.column")))
chrom28_narum_variants <- extract_variants_between_two_seqs(tq_seqs)
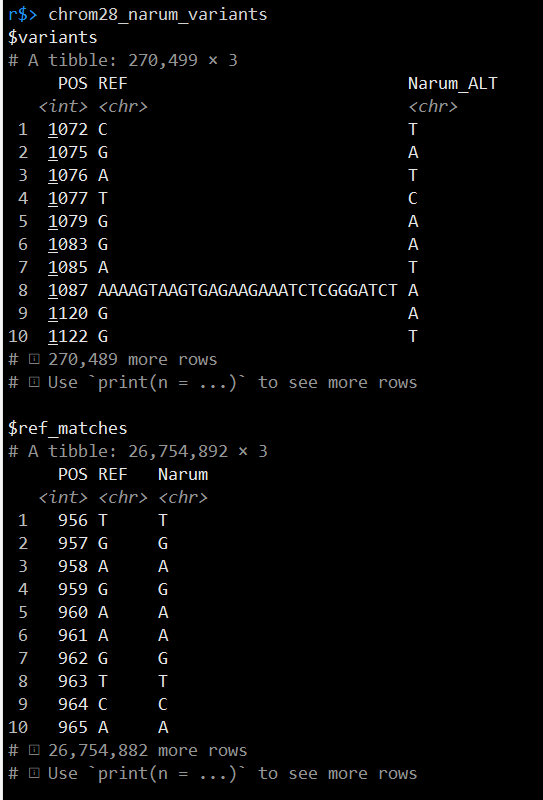
extract_variants_between_two_seqs.R 这个脚本的内容可以在 https://eriqande.github.io/thompson-et-al-2020-chinook-salmon-migration-timing/003-extract-johnson-creek-variants.html 链接处查看
欢迎大家关注我的公众号
小明的数据分析笔记本
小明的数据分析笔记本 公众号 主要分享:1、R语言和python做数据分析和数据可视化的简单小例子;2、园艺植物相关转录组学、基因组学、群体遗传学文献阅读笔记;3、生物信息学入门学习资料及自己的学习笔记!
| 




 苏公网安备32011502012024号
苏公网安备32011502012024号