VCF文件格式
VCF 是一种常用的生物信息学文件格式,用于**存储基因组中的遗传变异数据,特别是单核苷酸多态性(SNP)和小插入/缺失(Indel)**等变异。
vcftools软件安装
conda install vcftools -y
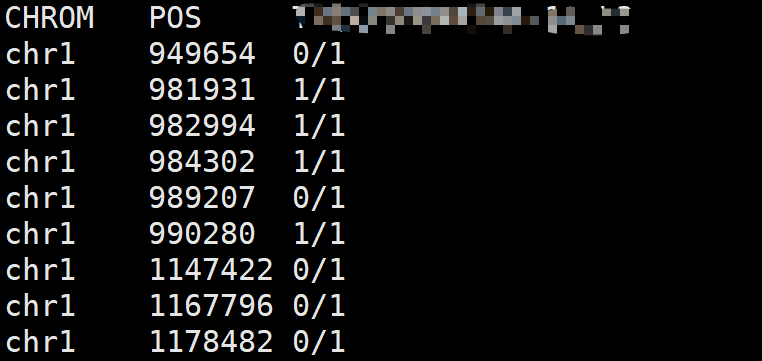
1. vcftools提取基因型
vcftools使用 --extract-FORMAT-info 选项来提取基因型信息。
vcftools --vcf sample.raw.vcf \
--extract-FORMAT-info GT --out sample
# 结果文件
# sample.GT.FORMAT
2. vcftools提取指定区域的变异信息
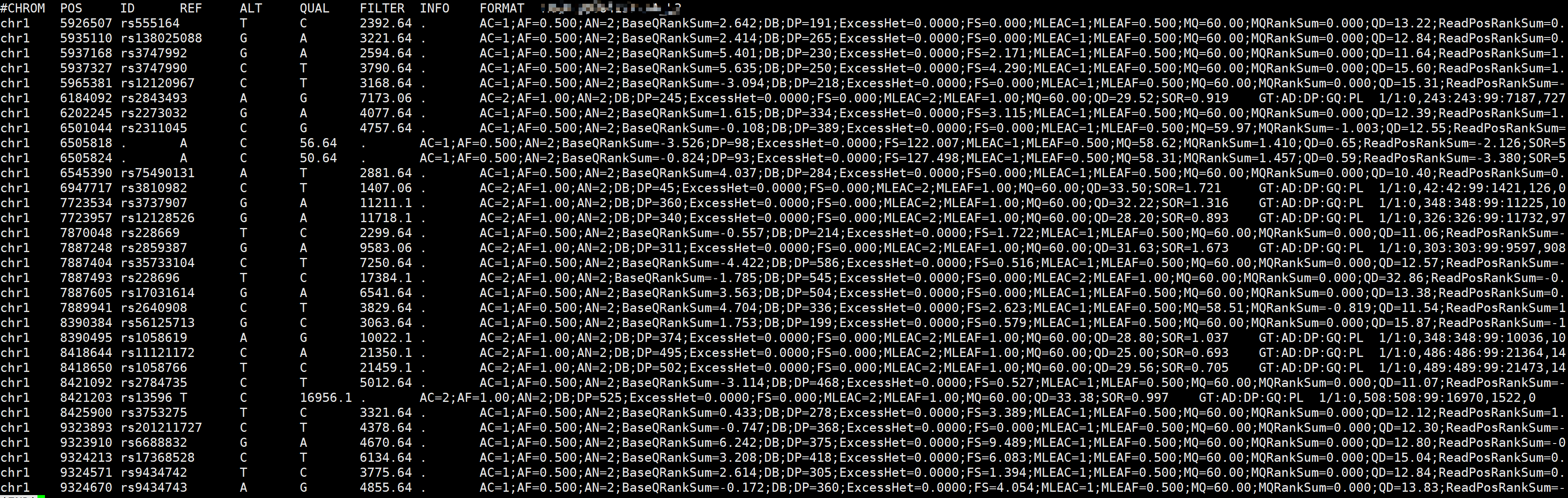
# 提取chr1 5M-10M区域的变异位点
vcftools --vcf sample.snp.vcf \
--chr chr1 --from-bp 5000000 --to-bp 10000000 \
--out sample --recode --recode-INFO-all
# 结果文件
# sample.recode.vcf
# 查看vcf结果文件
cat sample.recode.vcf|grep -v '^##'|less -S
参数说明:
--vcf :vcf文件 --chr :筛选区域所在的染色体 --form-bp :筛选区域的起始位置 --to-bp :筛选区域的终止位置 --out : 输出文件的前缀 --recode
3. vcftools对vcf文件进行划窗处理
对vcf文件进行滑窗处理,统计每个自定义大小窗口内的变异位点数量和多态性pi值。
# 对上述提取的sample.recode.vcf进行处理
vcftools --vcf sample.recode.vcf \
--window-pi 100 --window-pi-step 5
# 结果文件
# out.windowed.pi
参数说明:
--window-pi :指定窗口大小 --window-pi-step : 指定步长大小 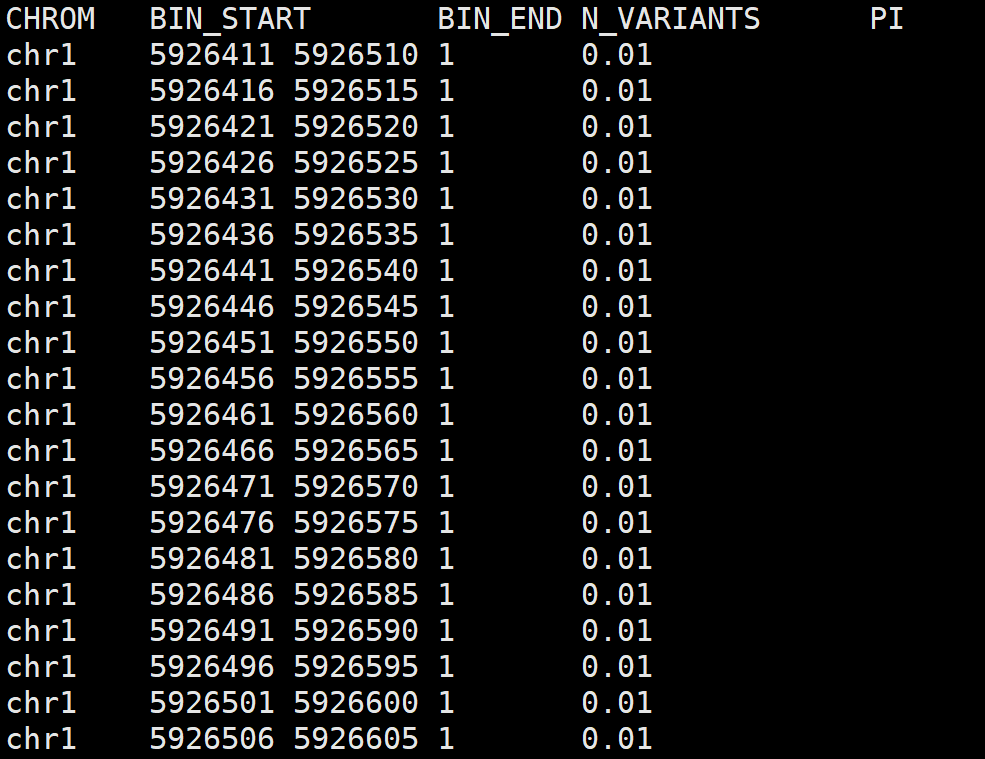
4. vcftools去除和保留vcf中指定样本
# 只保留sample01和sample02样本变异信息
vcftools --vcf sample.vcf --recode --recode-INFO-all \
--stdout --indv sample01 --indv sample2 > out.vcf
# 去除sample01样本变异信息
vcftools --vcf sample.vcf --recode --recode-INFO-all \
--stdout --remove-indv sample01 > out.vcf
# 根据文本文件保留样本变异信息
vcftools --vcf sample.vcf --recode --recode-INFO-all \
--stdout --keep sample_name.txt > out.vcf
# sample_name.txt内容
# sample01
# sample02
参数说明:
--keep <filename> : 保留样品 --remove <filename> : 删除样品
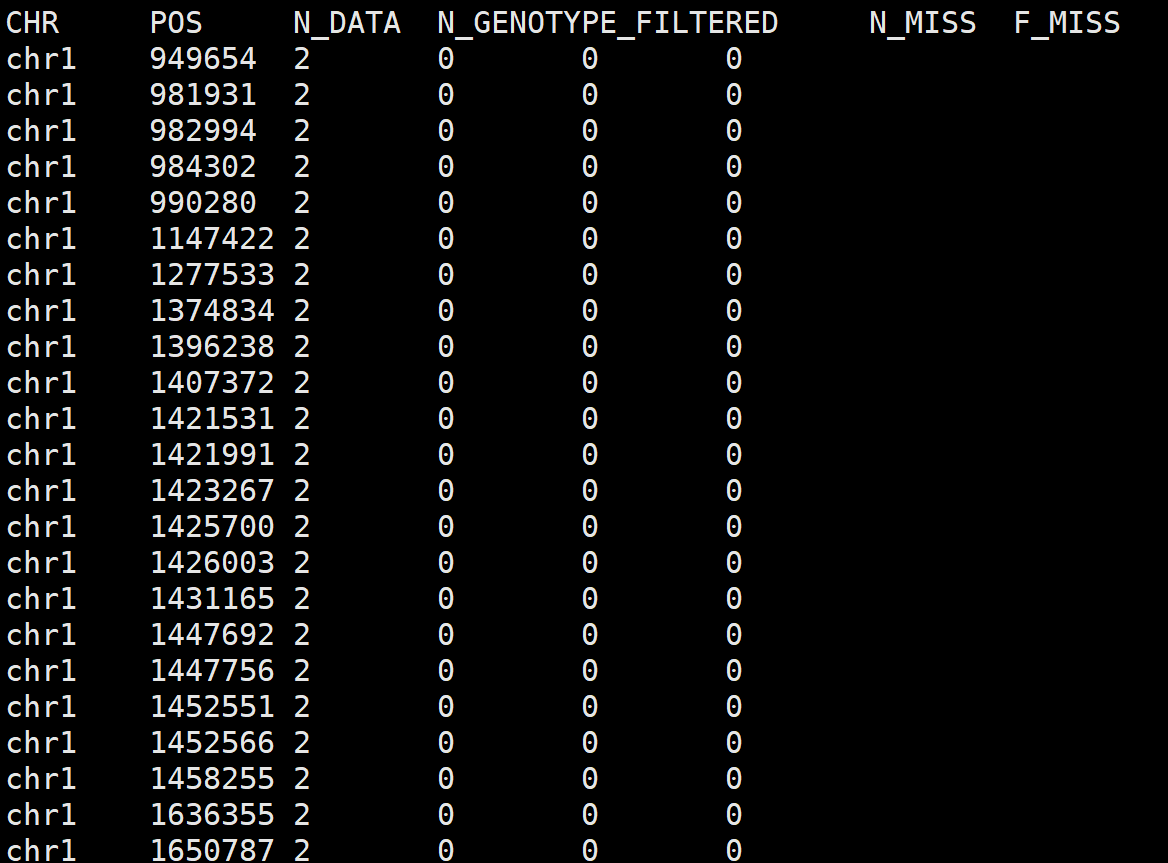
5. vcftools计算vcf文件的snp缺失率
vcftools --vcf sample.snp.vcf \
--missing-indv --out sample
# 结果文件
# sample.imiss
vcftools --vcf sample.snp.vcf \
--missing-site --out sample
# 结果文件
# sample.lmiss
参数说明:
--missing-indv :统计每个样品的缺失情况,生成文件后缀为“.imiss” --missing-site :统计每个snp位点的缺失情况,生成文件后缀为“.lmiss”
sample.imiss文件: 第1列为样品名称,第2列为总的snp数,第4列为缺失的总数,最后一列为缺失率。 
sample.lmiss文件:前2列为snp所在染色体和位置,第3列为等位基因总数,第5列为缺失的总数,最后一列为缺失率。
6. vcftools在重测序中应用过滤实例
vcftools --gzvcf sample.vcf.gz --recode --recode-INFO-all --stdout \
--maf 0.05 --max-missing 0.4 --minDP 4 --maxDP 1000 \
--minQ 30 --minGQ 80 --min-alleles 2 --max-alleles 2 |gzip - > sample.clean.vcf.gz






 苏公网安备32011502012024号
苏公网安备32011502012024号