bcftools可用于变异信息的描述性统计,计算,过滤和格式转换。
1. 显示VCF文件的头信息
bcftools view -h sample.vcf
##fileformat=VCFv4.2
##FILTER=<ID=PASS,Description="All filters passed">
##bcftoolsVersion=1.5+htslib-1.5
##bcftoolsCommand=mpileup -f /public/analysis/ucsc.hg19.fasta -Ou /public/analysis/result/sample.bam
##reference=file:///public/analysis/ucsc.hg19.fasta
##contig=<ID=chr1,length=249250621>
##contig=<ID=chr2,length=243199373>
##contig=<ID=chr3,length=198022430>
....
1. 构建VCF文件索引
# 对于分析依赖与vcf索引文件,进行以下操作生成.idx索引
bcftools index sample.vcf
bcftools index sample.vcf.gz
2. 压缩VCF为gz压缩文件
# 2线程输出.vcf.gz压缩文件
bcftools view sample.vcf -Oz -o sample.vcf.gz --threads 2
-Oz 表示输出格式为压缩文件gz格式; -o 后边跟压缩文件名字; –threads 2 表示2个线程并行压缩vcf文件
3. 压缩VCF为gz压缩文件
# 输出不压缩vcf
bcftools sort sample.vcf -o sample.vcf.gz -O v
# 输出压缩vcf.gz
bcftools sort sample.vcf -o sample.vcf.gz -O z
# -O 参数
# z: 压缩的VCF文件
# v: 不压缩的VCF文件
3. 提取VCF的等位基因和基因型信息
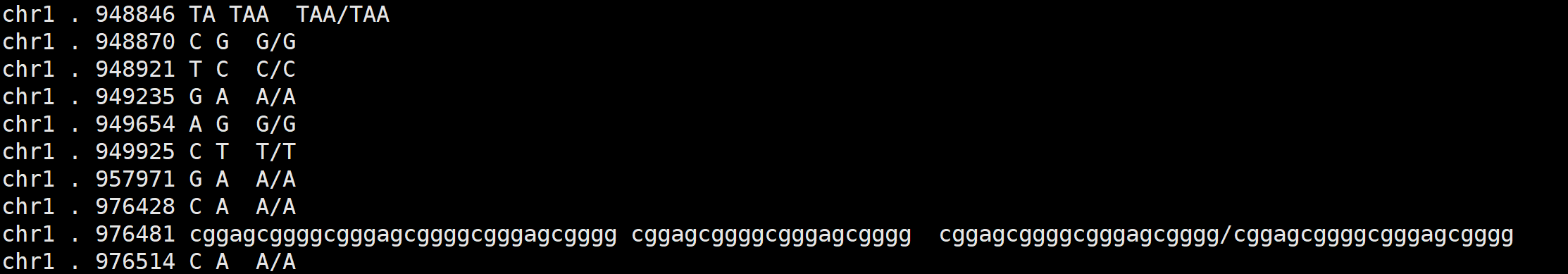
# 生成A/G的基因型
bcftools query -f '%CHROM %ID %POS %REF %ALT [ %TGT]\n' sample.vcf -o sample.extract.txt
%CHROM 染色体列
%ID 变异位点名称
%POS 变异位点位置
%REF 参考等位基因
%ALT 变异等位基因
%TGT 字符格式如A/G的基因型;%GT为0/1格式的基因型
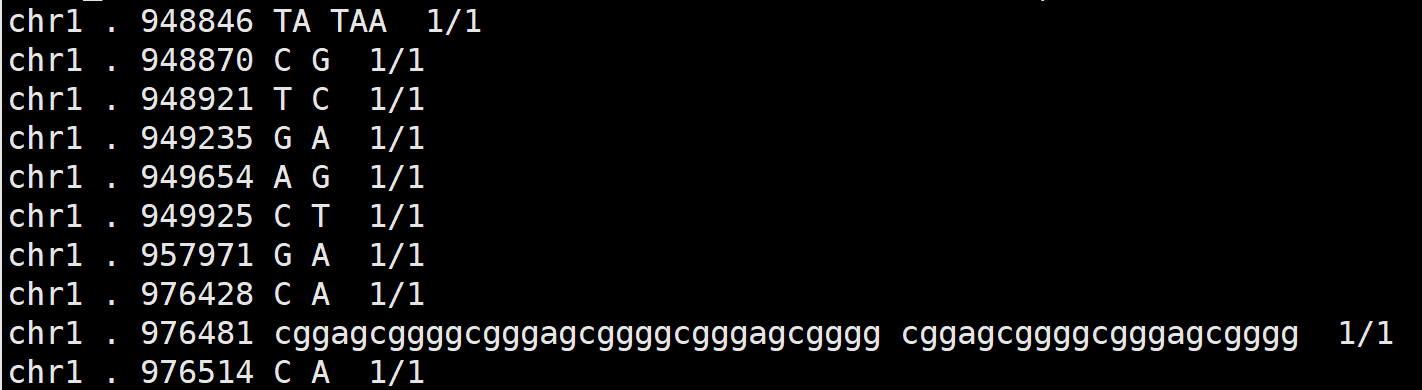
# 生成0/1格式的基因型
bcftools query -f '%CHROM %ID %POS %REF %ALT [ %GT]\n' sample.vcf -o sample.extract.txt
4. 变异位点的统计
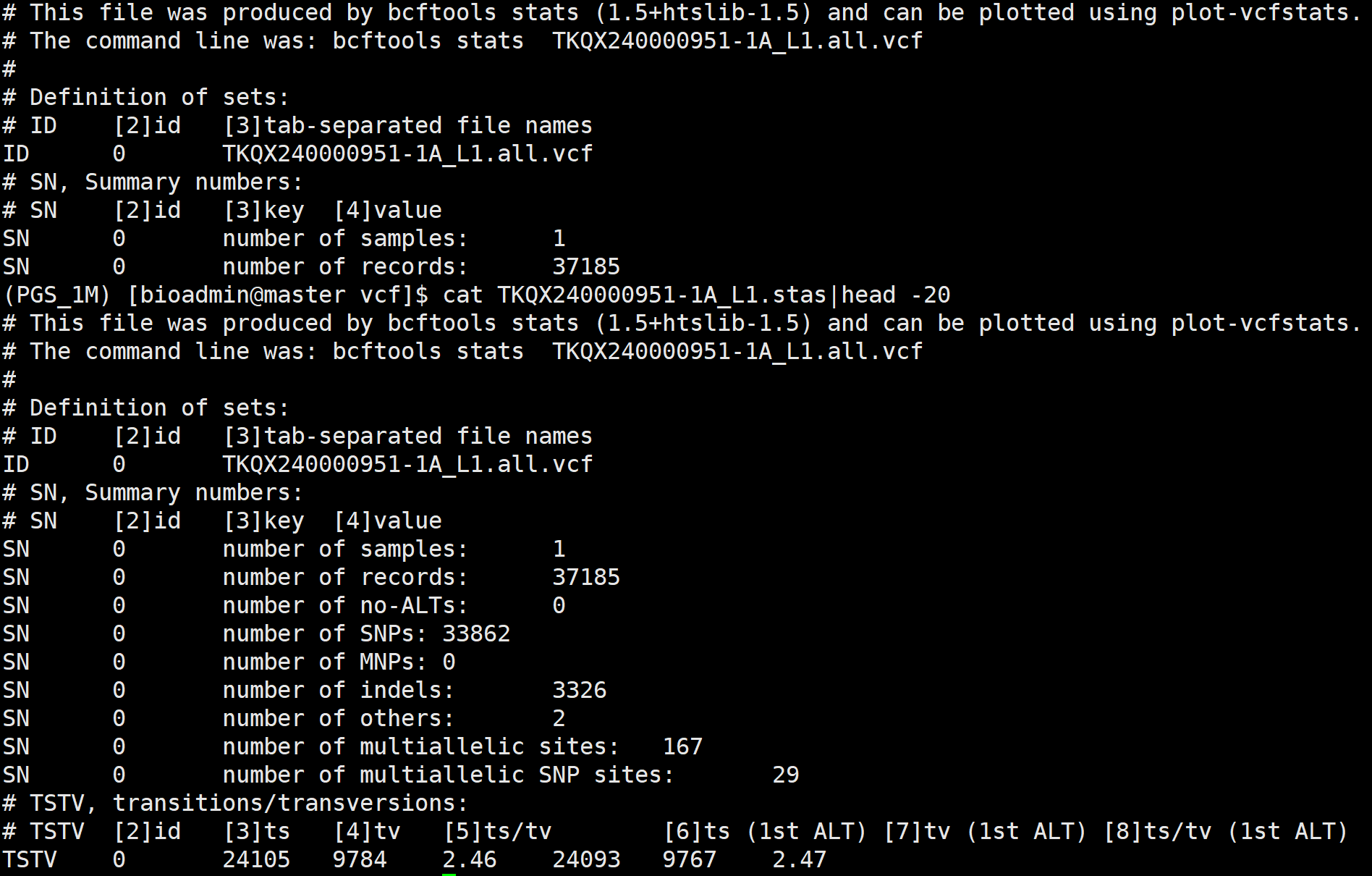
统计VCF文件的基本信息,比如突变位点的总数,不同类型突变位点的个数等。
# 统计命令
bcftools stats sample.vcf > sample.stas
# 安装plot-vcfstats 依赖库
# pip install matplotlib
# 生成pdf文件还需要pdf-latex, ubuntu使用下列命令安装
# sudo apt-get install texlive-full
# 统计可视化,输出值statistics文件夹
plot-vcfstats sample.stas -p statistics
5. 替换染色体名称
# 根据chr_name.txt文件替换染色体名称,并输出为.gz格式
bcftools annotate --rename-chrs chrom_name.txt old.vcf -Oz -o new.vcf.gz --threads 4
6. 使用数据库注释VCF文件
# 注释指定字段
## INFO/TAG可以写TAG
## FORMAT/TAG可以写FMT/TAG
################### dbSNP数据库注释 ######################
# 注释前需创建.vcf.gz文件索引, annoteate.vcf.gz 为注释压缩结果文件
bcftools index dbsnp.vcf.gz
# 使用dbSNP数据库vcf文件注释VCF的ID列, 获取rsid编号
bcftools annotate -a dbsnp.vcf.gz -c ID annoteate.vcf.gz
# gzip -d annoteate.vcf.gz
################### 1000G数据库注释 ######################
# 创建vcf文件index
bcftools index 1000g.vcf.gz
# 使用1000Gs数据库vcf文件注释VCF的等位基因频率AF列
bcftools annotate -a 1000g.vcf.gz -c AF annoteate.vcf.gz
# 使用1000Gs数据库vcf文件注释VCF的等位基因频率AF列和INFO列
bcftools annotate -a 1000g.vcf.gz -c INFO/AF annoteate.vcf.gz
bcftools annotate -a 1000g.vcf.gz -c INFO annoteate.vcf.gz






 苏公网安备32011502012024号
苏公网安备32011502012024号