ClassifyCNV是一个命令行工具,它实现了2019年ACMG指南,以评估生殖系重复和缺失。该工具使用预先解析的公开数据库来计算每个拷贝数变异的致病性得分 (CNV)。**
软件要求必须安装Python 3.6或以上版本和BEDTools 2.27.1或以上版本。
1. 获取github项目
git clone https://github.com/Genotek/ClassifyCNV.git
2. 输入文件: bed
ClassifyCNV接受BED文件作为输入, 支持hg19/hg38坐标。文件中必须包含以下列列信息:
chromosome染色体
CNV start positionCNV起始位置
CNV end positionCNV结束位置
CNV type (DEL or DUP)CNV类型(DEL或DUP)
ACMG_examples.hg19.bed文件示例:
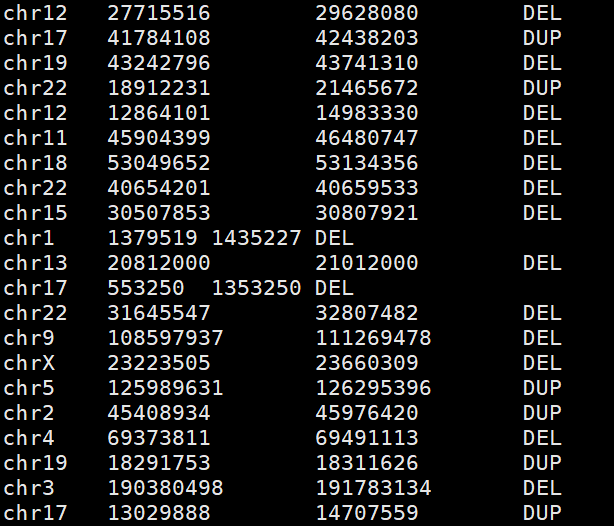
3. 运行示例
python ClassifyCNV.py --infile Examples/ACMG_examples.hg19.bed --GenomeBuild hg19 --precise
参数说明:
--infile: bed文件路径
--GenomeBuild: 参考基因组版本 hg19/hg38
-- cores: 要使用的线程数;默认值为1
--precise: 仅当确切的CNV断点已知时才应使用
--outdir: 指定的输出目录
通过ClassifyCNV计算的数值致病性评分使用 以下截止值:
≤ −0.99: benign variant
−0.90 - −0.98: likely benign variant
−0.89 - 0.89: variant of uncertain significance
0.90 - 0.98: likely pathogenic variant
≥ 0.99: pathogenic variant
≤ −0.99: 良性变体
−0.90 - −0.98: 可能是良性变异
−0.89 - 0.89: 不确定意义的变体
0.90 - 0.98: 可能的致病性变体
≥ 0.99:致病性变体
4. 输出注释结果
输出结果保存至ClassifyCNV_results文件夹中,结果文件夹以Result_dd_Mon_yyyy-hh-mm-ss格式命名, Scoresheet.txt为结果文件。文件的最后两列包括CNV中包含的剂量敏感基因的列表和CNV中所有蛋白质编码基因的列表。
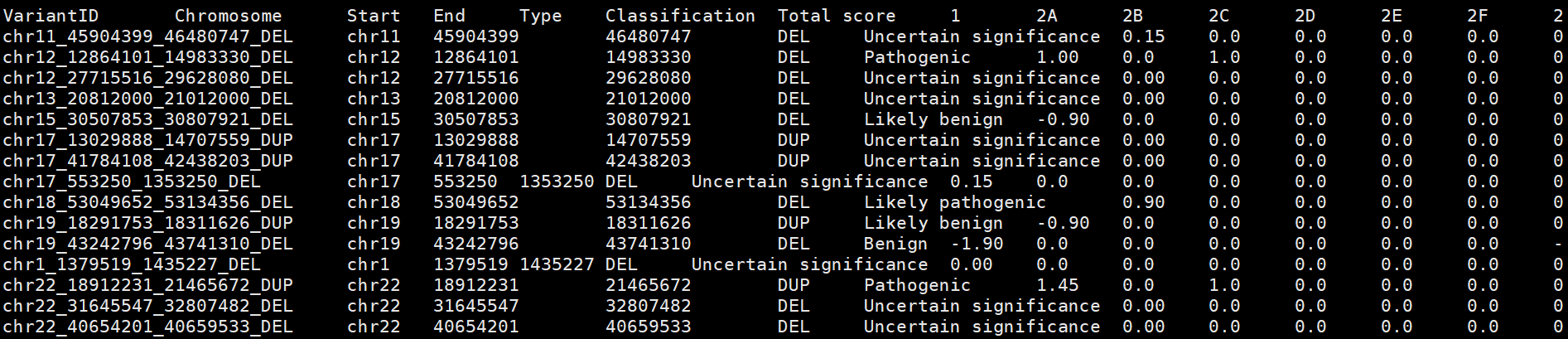
结果文件列名对应于ACMG规则的证据字段可在下列网址找到:
http://cnvcalc.clinicalgenome.org/cnvcalc/cnv-loss
http://cnvcalc.clinicalgenome.org/cnvcalc/cnv-gain
| 





 苏公网安备32011502012024号
苏公网安备32011502012024号