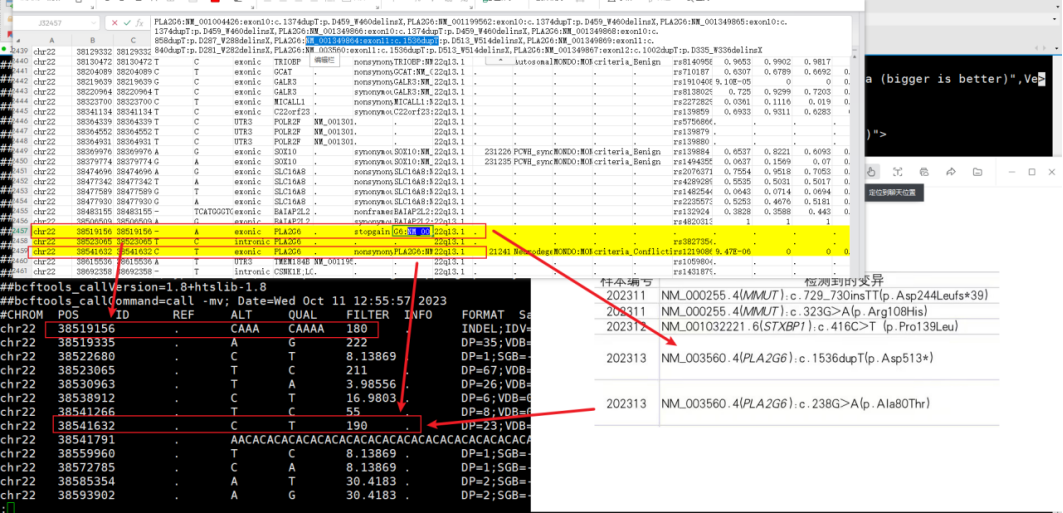
bcftools 作为实用的变异查找工具, 可以查找指定区域的变异; 相比于GATK可以互相补充。
1. conda 安装
conda install bcftools -y
2. bcftools 查找变异位点
# 查找chr22:38519150-385191650区域变异位点
bcftools mpileup -r chr22:38519150-385191650 \
-f /public/analysis/reference/hg19/hg19.fa \
-Ou Sample.sorted.bam |bcftools call -mv > chr22.vcf
3. bcftools 设置过滤参数查找变异位点
# 过滤比对质量小于10,碱基质量小于10
# FLAG为比对到基因组唯一位置UNMAP
bcftools mpileup -r chr22:38519150-385191650 \
-f /public/analysis/reference/hg19/hg19.fa \
-q 10 -Q 10 --ff UNMAP \
-Ou Sample.sorted.bam |bcftools call -mv > chr22.vcf
# 参数说明
-q, -min-MQ INT # 比对质量
Minimum mapping quality for an alignment to be used
-Q, --min-BQ INT # 碱基质量
Minimum base quality for a base to be considered
--ff, --excl-flags [UNMAP,SECONDARY,QCFAIL,DUP] # 根据FLAG筛选reads
详细参数见mpileup。 | 





 苏公网安备32011502012024号
苏公网安备32011502012024号