学习目标
- 了解如何在归一化过程中列出不同的
uninteresting factors(无关因素)
- 了解常用的归一化方法,已经如何使用
- 了解如何创建
DESeqDataSet 对象及其结构
- 了解如何使用
DESeq2 进行归一化
1. 归一化
差异表达分析工作流程的第一步是计数归一化,这是对样本之间的基因表达进行准确比较所必需的。
每个基因的映射读数计数是 RNA 表达以及许多其他因素的结果。归一化是调整原始计数值以解决“无关”因素的过程。以这种方式,表达水平在样本之间或样本内更具可比性。
在归一化过程中经常考虑的“无关”因素:
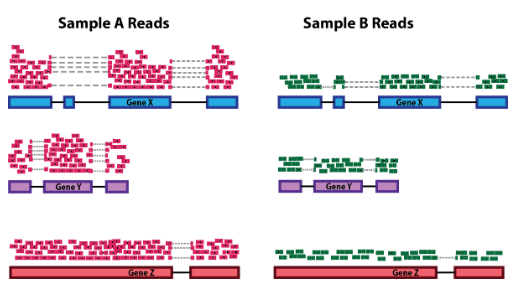
1.1. 测序深度
考虑测序深度对于比较样本之间的基因表达是必要的。在下面的示例中,每个基因在样本 A 中的表达似乎是样本 B 的两倍。然而,这是样本 A 的测序深度加倍的结果。
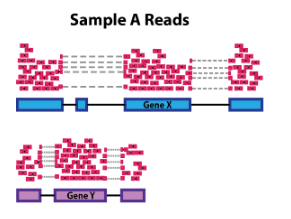
1.2. 基因长度
计算基因长度对于比较同一样本中不同基因之间的表达是必要的。在下面的示例中,基因 X 和基因 Y 具有相似的表达水平,但映射到基因 X 的读数数量将比映射到基因 Y 的读数多得多,因为基因 X 更长。
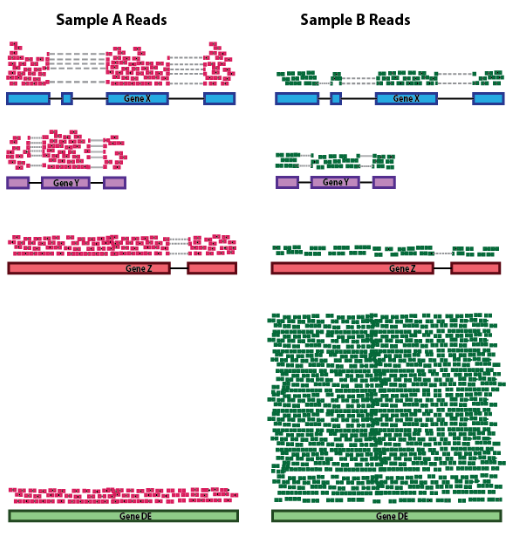
1.3. RNA组成
样本之间一些高度差异表达的基因、样本之间表达的基因数量的差异或污染的存在可能会扭曲某些类型的归一化方法。建议考虑 RNA 组成以准确比较样本之间的表达,这在进行差异表达分析时尤为重要。
在下面的示例中,假设样本 A 和样本 B 之间的测序深度相似,并且除了基因差异表达之外的每个基因在样本之间呈现相似的表达水平。样本 B 中的计数会受到 差异表达基因的极大影响,它占据了大部分计数。因此,样本 B 的其他基因的表达似乎低于样本 A 中的相同基因。
归一化不仅对于差异表达分析必不可少,对于探索数据分析、数据可视化以及探索或比较样本之间或样本内的计数也是必要的。
2. 归一化方法
几种常见的归一化方法:
| 方法 |
描述 |
考虑因素 |
使用场景 |
| CPM (counts per million) |
按照reads总数缩放计数 |
测序深度 |
同一样本组重复之间的基因计数比较;不适用于样本内比较或差异表达分析 |
| TPM (transcripts per kilobase million) |
每百万读取reads比对的转录本长度 (kb) 计数 |
测序深度与基因长度 |
样本内或同一样本组样本之间的基因计数比较;不适用于差异表达分析 |
| RPKM/FPKM (reads/fragments per kilobase of exon per million reads/fragments mapped) |
类似于TPM |
测序深度与基因长度 |
样本中基因之间的基因计数比较;不适用于样本比较或差异表达分析 |
| DESeq2’smedian of ratios |
计数除以特定于样本的大小因子,该因子由基因计数相对于每个基因的几何平均值的中位数比率确定 |
测序深度和RNA组成 |
样品之间的基因计数比较和差异表达分析;不适用于样本内比较 |
| EdgeR’strimmed mean of M values (TMM) |
使用样本之间对数表达比率的加权修剪平均值 |
测序深度和RNA组成 |
样品之间的基因计数比较和差异表达分析;不适用于样本内比较 |
虽然 TPM 和 RPKM/FPKM 归一化方法都考虑了测序深度和基因长度,但不推荐使用 RPKM/FPKM。原因是RPKM/FPKM方法输出的归一化计数值在样本之间没有可比性。
使用 RPKM/FPKM 归一化,每个样本的 RPKM/FPKM 归一化计数总数会有所不同。因此,您不能在样本之间平均比较每个基因的归一化计数。
RPKM-归一化计数表:
| gene |
sampleA |
sampleB |
| XCR1 |
5.5 |
5.5 |
| WASHC1 |
73.4 |
21.8 |
| … |
… |
… |
| Total RPKM-normalized counts |
1,000,000 |
1,500,000 |
例如,在上表中,样本 A 的 XCR1 (5.5/1,000,000) 计数比例高于样本 B (5.5/1,500,000),即使 RPKM 计数值相同。因此,我们不能直接比较样本 A 和样本 B 之间 XCR1(或任何其他基因)的计数,因为样本之间的归一化计数总数不同。
DESeq2-归一化计数:比率方法的中值(Median of ratios method)
由于用于差异表达分析的工具正在比较样本组之间相同基因的计数,因此该工具不需要考虑基因长度。然而,确实需要考虑测序深度和 RNA 组成。为了标准化测序深度和 RNA 组成,DESeq2 使用比率中值方法。在用户端只有一个步骤,但在后端涉及多个步骤,如下所述。
- 创建一个伪参考样本(逐行几何平均值)
对于每个基因,都会创建一个伪参考样本,该样本等于所有样本的几何平均值。
| gene |
sampleA |
sampleB |
pseudo-reference sample |
| EF2A |
1489 |
906 |
sqrt(1489 * 906) =1161.5 |
| ABCD1 |
22 |
13 |
sqrt(22 * 13) =17.7 |
| … |
… |
… |
… |
- 计算每个样本与参考的比率
对于每个样本中的每个基因,计算比率(样本/参考)(如下所示)。由于大多数基因没有差异表达,因此每个样本中的大多数基因在样本中的比例应该相似。
| gene |
sampleA |
sampleB |
pseudo-reference sample |
ratio of sampleA/ref |
ratio of sampleB/ref |
| EF2A |
1489 |
906 |
1161.5 |
1489/1161.5 =1.28 |
906/1161.5 =0.78 |
| ABCD1 |
22 |
13 |
16.9 |
22/16.9 =1.30 |
13/16.9 =0.77 |
| MEFV |
793 |
410 |
570.2 |
793/570.2 =1.39 |
410/570.2 =0.72 |
| BAG1 |
76 |
42 |
56.5 |
76/56.5 =1.35 |
42/56.5 =0.74 |
| MOV10 |
521 |
1196 |
883.7 |
521/883.7 =0.590 |
1196/883.7 =1.35 |
| … |
… |
… |
… |
|
|
- 计算每个样本的归一化因子(大小因子)
给定样本的所有比率的中值(上表中的列)被视为该样本的归一化因子(大小因子),计算如下。请注意,差异表达的基因不应影响中值:
normalization_factor_sampleA <- median(c(1.28, 1.3, 1.39, 1.35, 0.59))
normalization_factor_sampleB <- median(c(0.78, 0.77, 0.72, 0.74, 1.35))
下图说明了单个样本的所有基因比率分布的中值(y 轴是频率)。
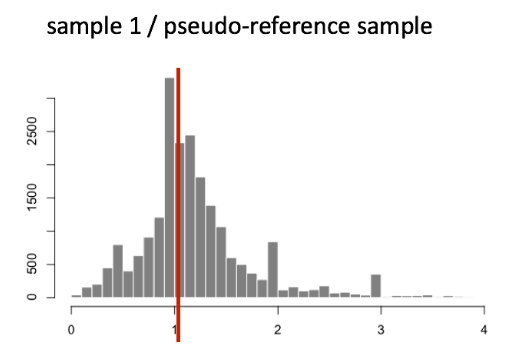
比率中位数法假设并非所有基因都差异表达;因此,归一化因子应考虑样本的测序深度和 RNA 组成(大的离群基因不会影响中值比率值)。该方法对上调/下调和大量差异表达基因的不平衡具有鲁棒性。
- 使用归一化因子计算归一化计数值
这是通过将给定样本中的每个原始计数值除以该样本的归一化因子来执行的,生成归一化计数值。这是针对所有计数值(每个样本中的每个基因)执行的。例如,如果样本 A 的中值比率为 1.3,样本 B 的中值比率为 0.77,则可以按如下方式计算归一化计数:
| gene |
sampleA |
sampleB |
| EF2A |
1489 |
906 |
| ABCD1 |
22 |
13 |
| … |
… |
… |
| gene |
sampleA |
sampleB |
| EF2A |
1489 / 1.3 =1145.39 |
906 / 0.77 =1176.62 |
| ABCD1 |
22 / 1.3 =16.92 |
13 / 0.77 =16.88 |
| … |
… |
… |
请注意,归一化计数值不是整数。
以上步骤仅作为演示,在实际使用DESeq2过程中,只需要一步命令,即可完成计算。
3. Mov10 归一化
现在我们知道了计数归一化的理论,我们将使用 DESeq2 对 Mov10 数据集的计数进行归一化。这需要几个步骤:
- 确保
metadata 数据框的行名存在,并且与 counts 数据框的列名顺序相同。
- 创建一个
DESeqDataSet 对象
- 生成归一化
counts
3.1. 数据匹配
我们应该始终确保样本名称在两个文件之间匹配,并且样本的顺序相同。如果不是这种情况,DESeq2 将输出错误。
# 检查两个文件中的样本名称是否匹配
all(colnames(txi$counts) %in% rownames(meta))
all(colnames(txi$counts) == rownames(meta))
如果数据不匹配,可以使用 match() 函数重新排列它们。
3.2. 创建对象
让我们从创建 DESeqDataSet 对象开始,然后可以更多地讨论其中存储的内容。要创建对象,我们需要将计数矩阵和元数据表作为输入。我们还需要指定一个设计公式。设计公式指定元数据表中的列以及它们在分析中的使用方式。对于我们的数据集,我们只有一列感兴趣,即 ~sampletype。此列具有三个因子水平,它告诉 DESeq2 对于每个基因,我们要评估相对于这些不同水平的基因表达变化。
我们的计数矩阵输入存储在 txi 列表对象中。所以我们需要指定使用 DESeqDataSetFromTximport() 函数,这将提取计数组件并将值四舍五入到最接近的整数。
# 对象创建
dds <- DESeqDataSetFromTximport(txi, colData = meta, design = ~ sampletype)
3.3. 生成归一化counts
下一步是对计数数据进行归一化,以便在样本之间进行正确的基因比较。
为了执行归一化比率方法的中位数,DESeq2 有一个 estimateSizeFactors() 函数可以生成大小因子。我们将在下面的示例中演示此功能,但在典型的 RNA-seq 分析中,此步骤由 DESeq() 函数自动执行,我们将在后面讨论。
dds <- estimateSizeFactors(dds)
通过将结果分配回 dds 对象,我们用适当的信息填充了 DESeqDataSet 对象。我们可以使用以下方法查看每个样本的归一化因子:
sizeFactors(dds)
现在,要从 dds 中检索归一化计数矩阵,我们使用 counts() 函数并添加参数 normalized=TRUE。
normalized_counts <- counts(dds, normalized=TRUE)
我们可以将这个归一化的数据矩阵保存到文件中以备后用:
write.table(normalized_counts, file="data/normalized_counts.txt", sep="\t", quote=F, col.names=NA)
注意:DESeq2 实际上并不使用归一化计数,而是使用原始计数并对广义线性模型 (GLM) 中的归一化进行建模。这些归一化计数对于结果的下游可视化很有用,但不能用作 DESeq2 或任何其他使用负二项式模型执行差异表达分析的工具的输入。








 苏公网安备32011502012024号
苏公网安备32011502012024号