 「一边学习,一边总结,一边分享!」
「一边学习,一边总结,一边分享!」
本期教程
本期教程,我们提供的原文的译文,若有需求请回复关键词:20240521
「小杜的生信笔记」,自2021年11月开始做的知识分享,主要内容是「R语言绘图教程」、「转录组上游分析」、**「转录组下游分析」等内容。凡是**在社群同学,可免费获得自2021年11月份至今全部教程,教程配备事例数据和相关代码,我们会持续更新中。
往期教程部分内容





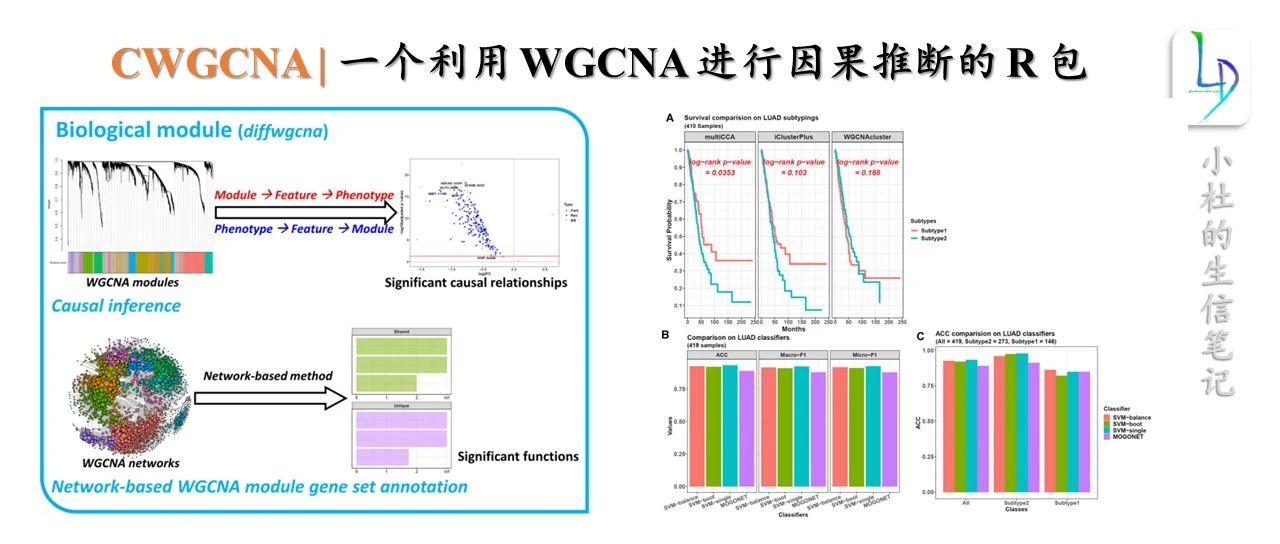
About CWGCNA
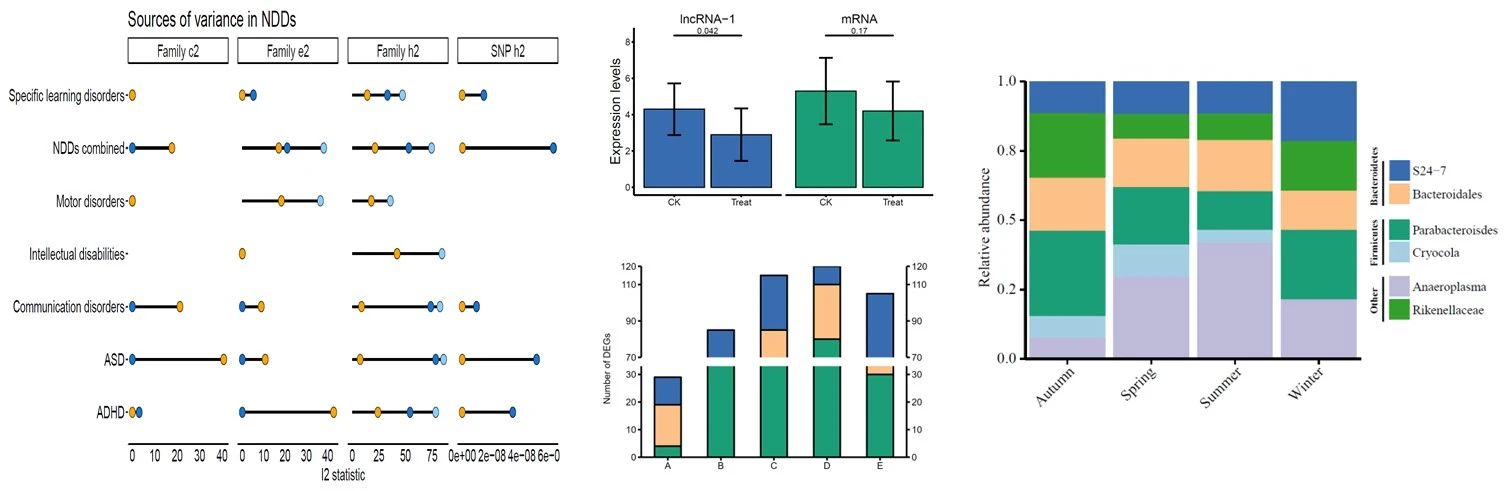
加权基因共表达网络分析(WGCNA)是鉴定共表达基因模块及其与表型性状相关性的有效工具。在这里,我们探索了更多的可能性,并开发了 R 包 CWGCNA (因果 WGCNA) ,它从传统的 WGCNA 管道工作,但挖掘更多的信息。它将一个中介模型与 WGCNA 结合起来,因此可以找到 WgcNA 模块、模块特征和表型之间的因果关系,证明模块的变化是否导致表型的变化,反之亦然。然后,在注释模块基因集函数时,考虑模块的拓扑结构,捕捉模块对基因集函数的影响,采用一种新的基于网络的方法。除了进行这些生物学探索,CWGCNA 还包含一个机器学习部分,对多组学数据进行聚类和分类,因为这种数据类型越来越受欢迎。一些基本功能,如差分特征识别,也可在我们的软件包。
Functional
- 方差分析(特征抽样)
- 因果WGCNA分析
- 多组学聚类(multiCCA和wgcnacluster)
- 多组学分类(组学分类器和配对组合预测)
- 多组学分类(组学分类器和配对组合预测)

发表单位:美国国家癌症研究所癌症研究中心病理学实验室 软件包网址:https://github.com/yuabrahamliu/CWGCNA/tree/v1.0.0
代码运行
- 安装
library(devtools)
install_github("yuabrahamliu/CWGCNA")
- 数据集
library(CWGCNA)
betas <- system.file("extdata", "placentabetas.rds", package = "CWGCNA")
betas <- readRDS(betas)
pds <- system.file("extdata", "placentapds.rds", package = "CWGCNA")
pds <- readRDS(pds)
#The DNAm betas matrix
betas[1:6,1:6]
#> GSM788417 GSM788419 GSM788420 GSM788421 GSM788414 GSM788415
#> cg00000292 0.65961366 0.67591141 0.65709651 0.66077820 0.66847653 0.67436406
#> cg00002426 0.53516824 0.53883284 0.53683120 0.53990206 0.53804637 0.53543344
#> cg00003994 0.17674229 0.16432771 0.16631494 0.16803355 0.16416337 0.16852233
#> cg00007981 0.03553198 0.02934814 0.03189098 0.02765783 0.02768899 0.02764183
#> cg00008493 0.43619912 0.42998023 0.43761396 0.44973257 0.46281094 0.45935308
#> cg00008713 0.07431056 0.06367040 0.06764089 0.06218142 0.05267028 0.05397539
#The metadata for the samples
head(pds)
#> sampleid Group Gestwk Babygender Ethnicity Dataset
#> 1 GSM788417 Control 8 M White GSE31781
#> 2 GSM788419 Control 8 M White GSE31781
#> 3 GSM788420 Control 8 M White GSE31781
#> 4 GSM788421 Control 9 M White GSE31781
#> 5 GSM788414 Control 12 F Asian GSE31781
#> 6 GSM788415 Control 12 M White GSE31781
table(pds$Group)
#>
#> Control Preeclampsia
#> 258 101
table(pds$Dataset)
#>
#> GSE100197 GSE125605 GSE31781 GSE36829 GSE59274 GSE69502 GSE73375 GSE74738
#> 65 41 30 48 23 16 36 28
#> GSE75196 GSE98224
#> 24 48
- Confounding factor analysis
#This command will take < 1 min
top10k <- featuresampling(betas = betas,
topfeatures = 10000,
variancetype = "sd",
threads = 6)
top10k$betas[1:6,1:6]
#> GSM788417 GSM788419 GSM788420 GSM788421 GSM788414 GSM788415
#> cg00000292 0.6596137 0.6759114 0.6570965 0.6607782 0.6684765 0.6743641
#> cg00002426 0.5351682 0.5388328 0.5368312 0.5399021 0.5380464 0.5354334
#> cg00003994 0.1767423 0.1643277 0.1663149 0.1680336 0.1641634 0.1685223
#> cg00008493 0.4361991 0.4299802 0.4376140 0.4497326 0.4628109 0.4593531
#> cg00013618 0.7859429 0.7868591 0.7933922 0.7958716 0.8006630 0.8118193
#> cg00014837 0.8522323 0.8525327 0.8516924 0.8546466 0.8610099 0.8546916
dim(top10k$betas)
#> [1] 10000 359
head(top10k$varicanceres)
#> SD
#> cg03729431 0.1848922
#> cg16063666 0.1643842
#> cg05790038 0.1456048
#> cg03316864 0.1454261
#> cg11009736 0.1393115
#> cg20322876 0.1348288
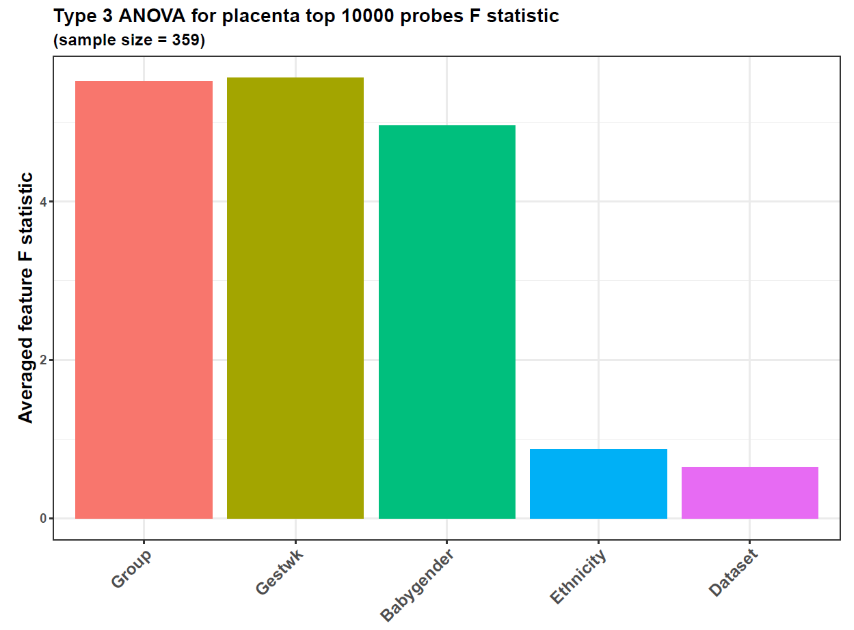
#This command will take < 1 min
anovares <- featuresampling(betas = top10k$betas,
pddat = pds,
anova = TRUE,
plotannovares = TRUE,
featuretype = "probe",
plottitlesuffix = "placenta",
titlesize = 18,
textsize = 16,
threads = 6)
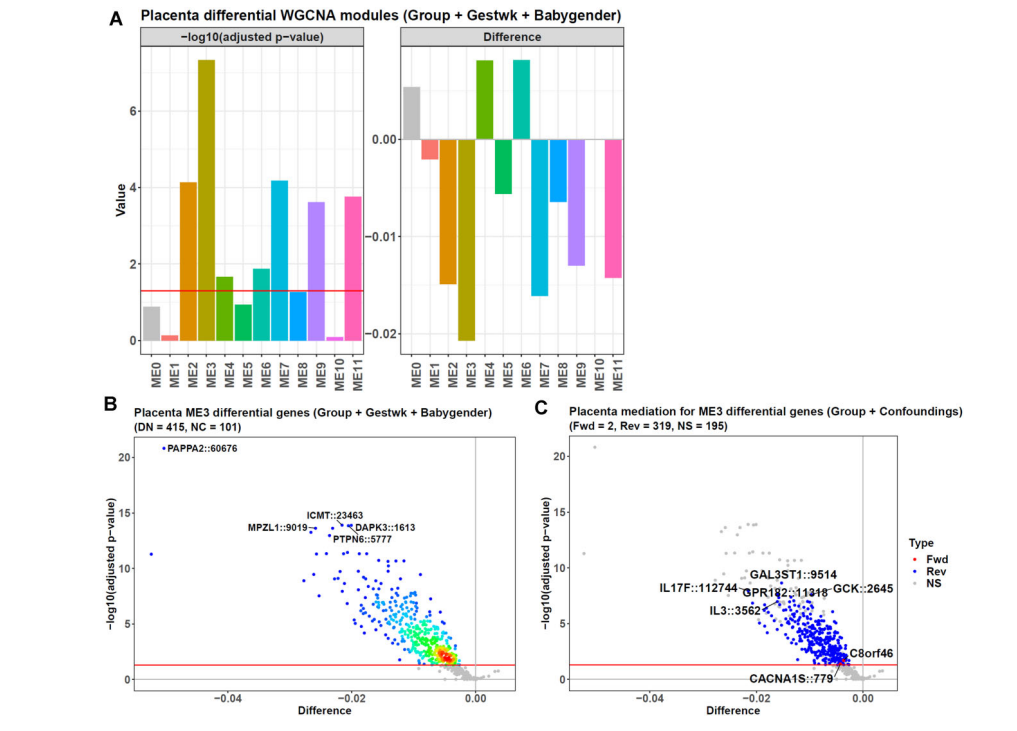
- Causal WGCNA analysis
#This command will take < 1 min
betasgene <- probestogenes(betadat = betas, group450k850k = c("TSS200", "TSS1500", "1stExon"))
This WGCNA analysis is time-consuming (~ 25 min), and the running below can be skipped.
#This command will take ~ 25 min
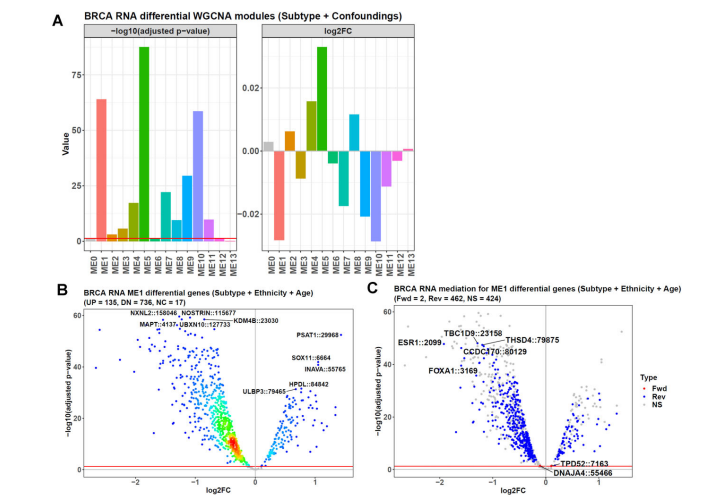
cwgcnares <- diffwgcna(dat = betasgene,
pddat = pds,
responsevarname = "Group",
confoundings = c("Gestwk", "Babygender"),
featuretype = "gene",
topvaricancetype = "sd",
topvaricance = 5000,
powers = seq(1, 20, 1),
rsqcutline = 0.8,
mediation = TRUE,
topn = 1,
plot = TRUE,
titleprefix = "Placenta",
labelnum = 5,
titlesize = 17,
textsize = 16,
annotextsize = 5,
pvalcolname = "adj.P.Val",
pvalcutoff = 0.05,
isbetaval = TRUE,
absxcutoff = 0,
diffcutoff = 0,
threads = 6)
- PCA/CCA-based clustering and bagging-SMOTE classification
#Extract the DNAm probe data for the 101 preeclampsia samples
prepds <- subset(pds, Group == "Preeclampsia")
row.names(prepds) <- 1:nrow(prepds)
prebetas <- betas[, prepds$sampleid, drop = FALSE]
#Clustering
#This command will take < 1 min
presubtyperes <- multiCCA(dats = list(prebetas),
k = 2,
consensus = 1,
seednum = 2022,
threads = 6,
plot = TRUE,
titlefix = "Preeclampsia",
titlesize = 18,
textsize = 16)
 The result presubtyperes is a list, and its slot "ivis" records 3 internal validation indices for each candidate cluster number.
The result presubtyperes is a list, and its slot "ivis" records 3 internal validation indices for each candidate cluster number.
presubtyperes$ivis
#> $`k = 2`
#> silhouette calinski_harabasz davies_bouldin
#> 0.2622399 36.6521374 1.3709824
The slot "kreses" contains the cluster labels for samples, and its sub-slot "k = 2" is for the cluster number of 2.
head(presubtyperes$kreses$`k = 2`)
#> GSM1892055 GSM2674426 GSM2674432 GSM2589560 GSM2674433 GSM1892048
#> 2 1 2 2 2 1
table(presubtyperes$kreses$`k = 2`)
#>
#> 1 2
#> 29 72
In addition to balancing sample distribution, the bagging-SMOTE framework also performs feature selection with elastic net. Then, it uses these features to construct several SVM (support vector machine) or MLR (multinomial logistic regression) base learners and finally ensembles their results.
#Make the sample labels from the clustering result
subtypes <- paste0("Subtype", presubtyperes$kreses$`k = 2`)
#Classification
#This command will take < 5 min
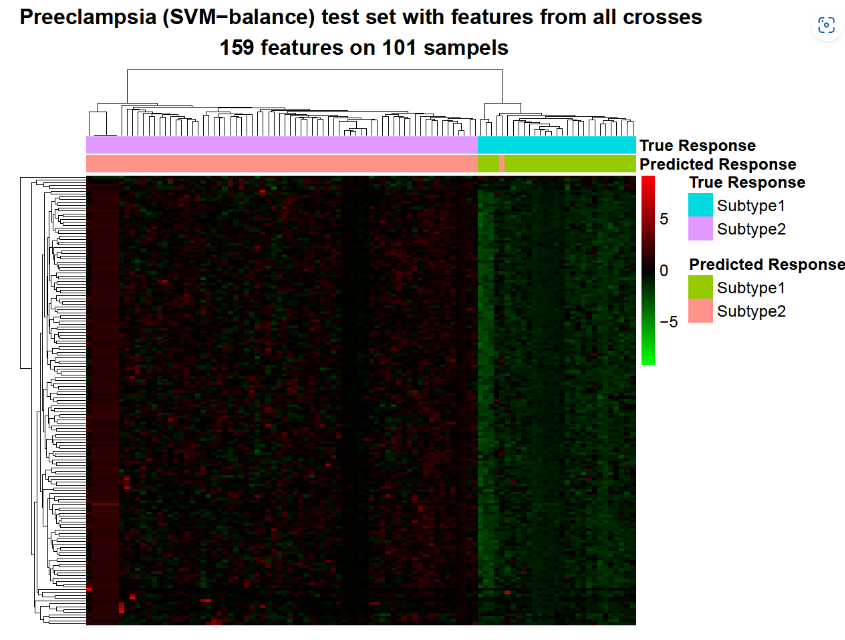
presubtypeclassifierres <- omicsclassifier(dats = list(prebetas),
truelabels = subtypes,
balanceadj = 1,
method = "SVM",
alphas = c(0.5),
nfold = 5,
seednum = 2022,
threads = 6,
plot = TRUE,
prefixes = c("Preeclampsia (SVM-balance)"))
软件包网址:https://github.com/yuabrahamliu/CWGCNA/tree/v1.0.0
本期教程,我们提供的原文的译文,若有需求请回复关键词:20240521
❝
若我们的教程对你有所帮助,请点赞+收藏+转发,这是对我们最大的支持。
❞
往期部分文章
「1. 最全WGCNA教程(替换数据即可出全部结果与图形)」
「2. 精美图形绘制教程」
「3. 转录组分析教程」
「4. 转录组下游分析」
「小杜的生信筆記」 ,主要发表或收录生物信息学教程,以及基于R分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!






 苏公网安备32011502012024号
苏公网安备32011502012024号