摘要
所有系统发育推断方法都需要同源数据集作为输入。因此,当核苷酸序列用于系统发育分析时,第一步通常是推断不同类群序列中的哪些核苷酸彼此同源,以便这些核苷酸之间的差异仅源于序列进化中发生的变化。不同序列的核苷酸之间的同源性推断最常通过属于“多序列比对”类别的方法来完成。
在本教程中,我将介绍如何使用最快、最流行的多序列比对工具之一,程序 MAFFT(Katoh 和 Standley 2013)。我将进一步演示如何检测和排除其中核苷酸同源性可能存在问题的比对区域,如何使用公共序列数据库(NCBI 的 GenBank)识别其他同源序列,以及如何使用这些序列来补充现有数据集。
数据集
本教程中使用的数据集是 Matschiner 等人使用的数据的一小部分。估计非洲和新热带丽鱼科鱼类与冈瓦纳大陆印度、马达加斯加、非洲和南美洲分裂相关的分化时间。这里使用的数据集包括两个基因的序列;编码 16S 核糖体 RNA 的线粒体 16S 基因和编码重组激活蛋白 1 的核 RAG1 基因。
依赖
- MAFFT:MAFFT 网页上提供了 MAFFT 的安装说明和预编译版本。虽然该程序的安装在所有操作系统上都应该很容易,但本教程的所有步骤也可以使用 MAFFT 的服务器版本进行;因此,该软件的安装是可选的。
- AliView:为了可视化序列比对,推荐使用软件 AliView (Larsson 2014)。 AliView 的安装在 http://www.ormbunkar.se/aliview/ 中进行了描述,并且应该可以在所有操作系统上进行。
- BMGE:BMGE对于识别和删除序列比对中对齐不良的区域非常有用。最新版本的 BMGE 以 Java jar 文件形式提供,位于 ftp://ftp.pasteur.fr/pub/gensoft/projects/BMGE/。
比对与可视化
我们将首先使用 MAFFT 程序比对线粒体 16S 基因的序列,然后使用软件 AliView 可视化并改进比对。
- 将包含 16S 序列的文件
16s.fasta 下载到您的分析目录。在文本编辑器或命令行上查看该文件,例如使用 less 命令:
less 16s.fasta
您将看到每条记录都由一个 ID 和一个序列组成,其中 ID 始终位于以“>”符号开头的单行上,后面是包含序列的行。序列尚未对齐;这就是它们不包含间隙且长度不同的原因。可以应用其他命名方案,而不是该文件中使用的 14 个字符的 ID;但是,我强烈建议使用简短的 ID,因为在系统发育分析中,如果您使用包含空格或连字符的实际拉丁名或常见物种名称,许多程序或脚本可能无法工作。
- 打开 MAFFT 在线版本的网站。该网站提供了 MAFFT 对齐程序的 Web 界面。如果您成功安装了 MAFFT,您还可以在计算机上使用 MAFFT,而不是使用该网站。
- 在 MAFFT 服务器网站上的“高级设置”标题下(向下滚动查看),您将找到可用的对齐选项。在第一个标题为“策略”的灰色框中,您可以在全局和局部对齐方法之间进行选择。 “G-INS-i”方法实现全局 Needleman-Wunsch 算法(Needleman 和 Wunsch 1970),“L-INS-i”方法实现局部“Smith-Waterman”算法(Smith 和 Waterman 1981)。为简单起见,保留默认的“自动”选项。如果您在自己的计算机上使用 MAFFT 的命令行版本而不是 MAFFT 服务器,则等效命令如下:
mafft --auto 16s.fasta > 16s_aln.fasta
- 在“高级设置”部分的第三个灰色框中,标题为“参数”,您可以更改评分矩阵。对于氨基酸序列,您可以选择任何与 PAM 矩阵等效的 BLOSUM 矩阵。对于核苷酸序列,可以选择“1PAM / K=2”、“20PAM / K=2”和“200PAM / K=2”。目前,保留所有默认选项。单击“提交”按钮。将 Fasta 格式的比对下载到您的计算机。为此,请右键单击页面最顶部的“Fasta 格式”链接。将文件命名为 16s_aln.fasta。
- 重复相同的操作,这次惩罚设置为 2,而不是默认值 1.53。将分析所得的比对文件命名为 16s_op2_aln.fasta。如果您使用 MAFFT 的命令行版本,则等效命令如下:
mafft --auto --op 2 16s.fasta > 16s_op2_aln.fasta
- 在AliView中打开文件16s_aln.fasta。在不关闭 AliView 窗口的情况下,在第二个 AliView 窗口中打开文件 16s_op2_aln.fasta。比较右下角状态栏中显示的总对齐长度。在两个 AliView 窗口中,滚动到位置 1250 和 1350 之间的区域。
- 在 16s_aln.fasta 的窗口中,识别对齐不良的区域(例如位置 1020 到 1040 周围)并尝试重新对齐。为此,请通过单击路线顶部的标尺来选择区域,如下面的屏幕截图所示。
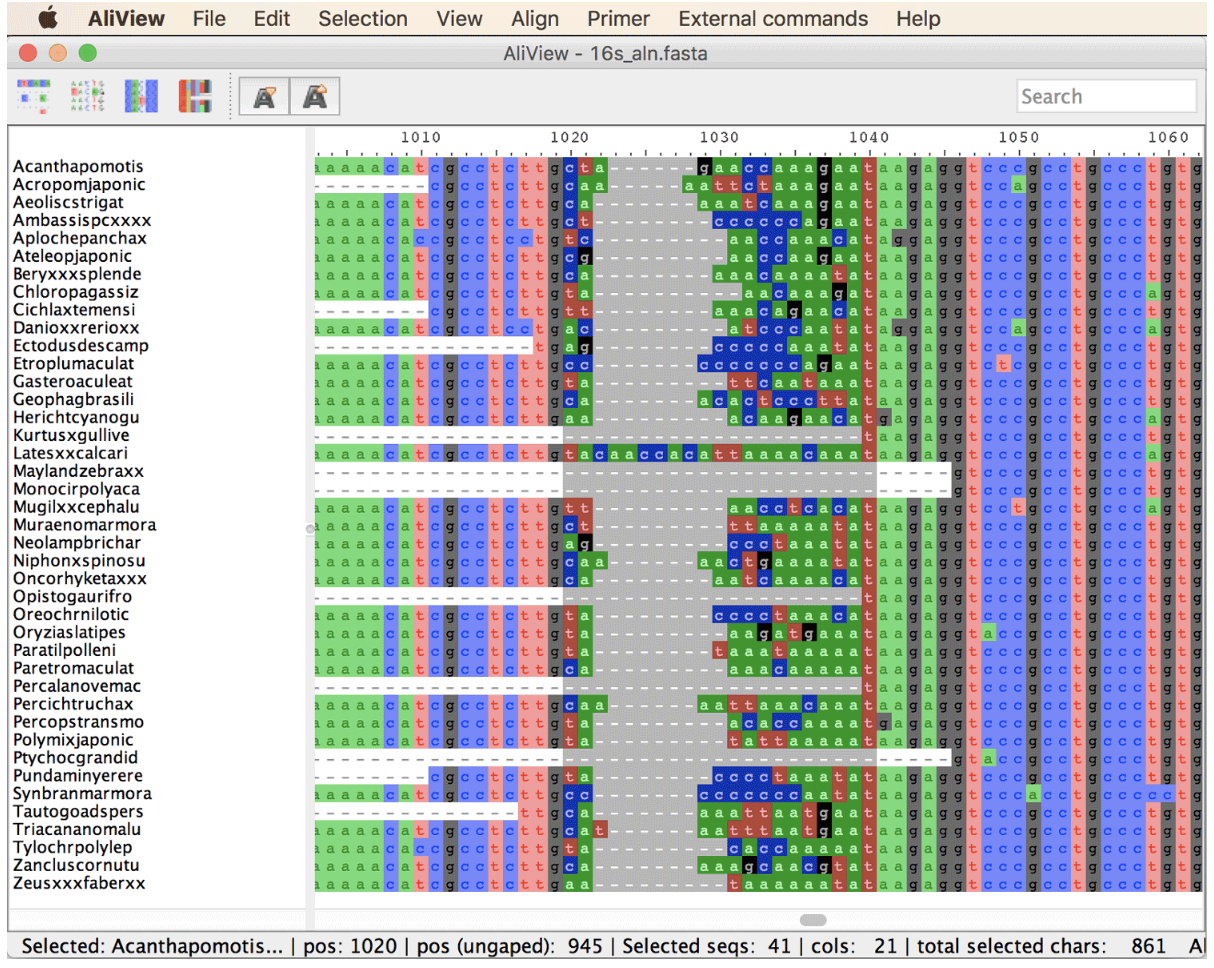
- 选择对齐不良的区域后,单击 AliView 的“对齐”菜单中的“重新对齐所选块”。
BMGE 自动对齐过滤
正如您所看到的,16S 序列的比对包含高度可变区域和保守区域的混合。因此,核苷酸的同源性在基因的某些部分相当明显,但在其他部分可能不明确。为了避免下游系统发育分析中的比对错误导致的问题,我们将根据缺口的比例和这些区域内发现的遗传变异来识别比对不良的区域,并将它们从比对中排除。
- 要从 16S 比对中排除不可靠的比对区域,请使用软件 BMGE。要检查该程序是否在您的计算机上运行并查看可用选项,请打开命令行窗口(例如 Mac OSX 上的终端应用程序)并键入以下命令:
java -jar BMGE.jar -?
# 如果上述方法有效,请输入以下命令:
java -jar BMGE.jar -i 16s_aln.fasta -t DNA -of 16s_filtered.fasta -oh 16s_filtered.html
通过上述命令,BMGE 以 Fasta 格式在文件 16s_filtered.fasta 中写入过滤后的比对,并在文件 16s_filtered.html 中以 HTML 格式可视化过滤后的比对。在浏览器中打开文件 16s_filtered.html。滚动浏览对齐并注意黑色对齐块。在对齐的最顶部,您将看到为每个站点以浅灰色和黑色绘制的两个值。差距比例用浅灰色等号显示,范围从 0 到 1。黑色冒号表示 BMGE 的作者所说的“平滑熵状分数”(Criscuolo 和 Gribaldo 2010)。基本上,这是对该位点核苷酸多样性的衡量。您会注意到黑色对齐块与低间隙比例和低熵的区域一致,这是最适合系统发育推断的对齐位置。我们对对齐块的选择基于 BMGE 的熵分数截止(选项 -h)、间隙率截止(-g)和最小块大小(-b)的默认设置。默认情况下,BMGE 选择熵分数低于 0.5 (-h 0.5) 且间隙比例低于 0.2 (-g 0.2) 的位点,并且仅当这些位点形成至少 5 个具有这些属性的位点 (-b 5) 时。
- 使用熵分数截止、间隙率截止和最小块大小的自定义设置重复 BMGE 块选择,并注意这如何改变所选站点的总数以及对齐中所选块的分布。例如,使用 -g 0.3 增加允许的间隙比例:
java -jar BMGE.jar -i 16s_aln.fasta -t DNA -g 0.3 -of 16s_g03_filtered.fasta -oh 16s_g03_filtered.html
- BMGE 到终端的标准输出告诉您有多少站点(字符)仍被选中。请注意最后两次运行之间的差异。除了文件 16s_filtered.html 之外,还要在单独的浏览器窗口中打开文件 16s_g03_filtered.html。滚动对齐。您会注意到,由于每个站点允许的间隙比例增加,现在有更多区域被标记为黑色。
- 在AliView中打开文件16s_filtered.fasta。请注意,它现在比以前的对齐方式更短并且看起来更压缩。使用 AliView 的“文件”菜单中的“另存为 Phylip(全名和填充)”选项,将文件以 Phylip 格式保存为 16s_filtered.phy。还可以使用“另存为 Nexus”选项将文件保存为 Nexus 格式的 16s_filtered.nex。
- 在文本编辑器中打开 Phylip 和 Nexus 文件以查看文件格式之间的差异。
| 







 苏公网安备32011502012024号
苏公网安备32011502012024号