动动发财的小手,点个赞吧!
简介
ABBA BABA 统计(也称为 D 统计)为偏离严格的分叉进化历史提供了简单而有力的检验。因此,它们经常用于使用基因组规模的 SNP 数据测试基因渗入。
虽然最初开发用于基因渗入的全基因组测试,但它们也可以应用于较小的窗口,从而可以探索基因渗入的基因组景观。
在本次实践中,我们将使用可用的软件执行基于窗口的 ABBA BABA 分析,然后在 R 中编写代码来绘制结果。我们将分析几个 Heliconius 蝴蝶种群的基因组数据。
工作流程
从多个个体的全基因组测序的基因型数据开始,我们将运行一个脚本来计算每条染色体上各个窗口中的混合比例的度量。然后我们将绘制图表来测试有关适应性基因渗入的假设。
数据集
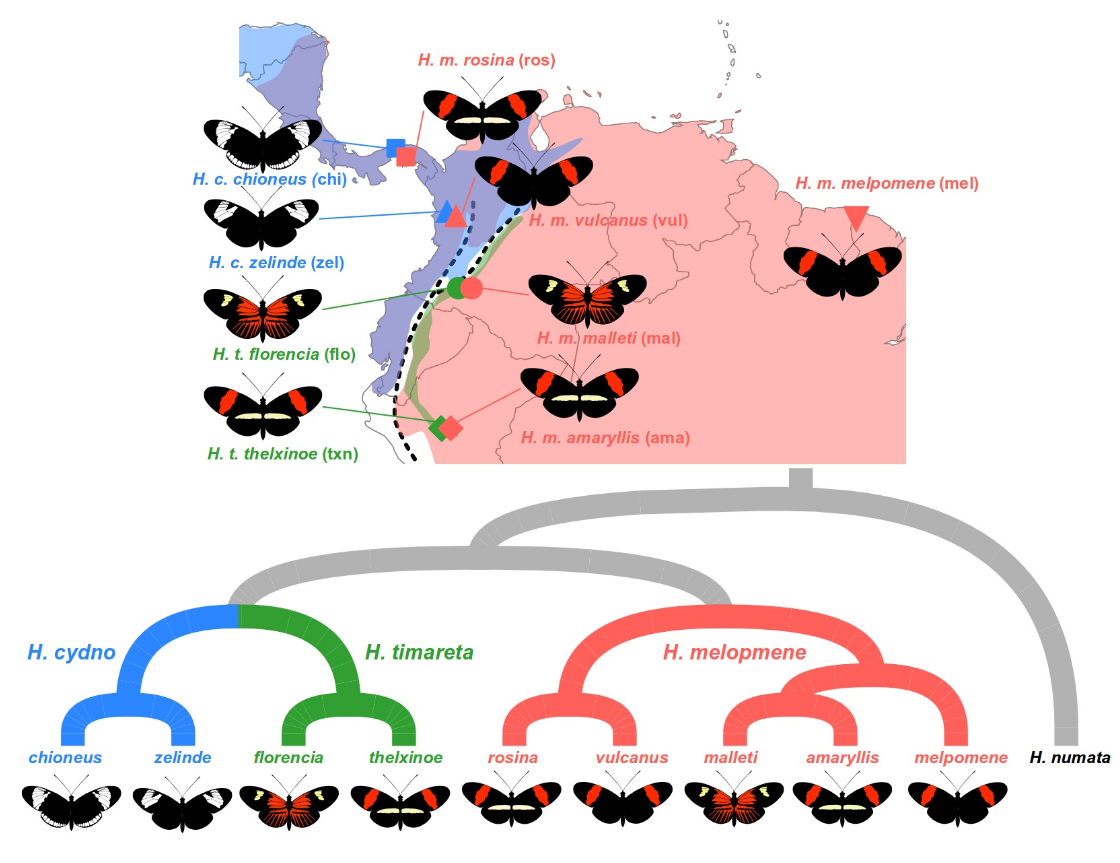
我们将研究三个物种的多个种族:Heliconius melpomene、Heliconius timareta 和 Heliconius cydno。这些物种的分布范围部分重叠,人们认为它们在同源地区发生杂交。我们的样本集包括来自巴拿马和哥伦比亚安第斯山脉西坡的两对同域种群 H. melpomene 和 H. cydno。在哥伦比亚和秘鲁的安第斯山脉东坡,还有两对同域种群:H. melpomene 和 H. timareta。最后,有两个来自外群物种 Heliconius numata 的样本,这是进行 ABBA BABA 分析所必需的。
所有样本均使用深度全基因组测序进行测序,并使用标准流程为每个个体的基因组中每个位点获取基因型。数据经过过滤,仅保留双等位基因单核苷酸多态性 (SNP)。该数据集包括来自 18 号染色体的 SNP 数据,已知该染色体携带特别感兴趣的翅膀图案基因座。
假设
我们假设同域物种之间的杂交将导致 H. cydno 和来自西方的 H. melpomene 同域种族之间以及 H. timareta 和来自东部的 H. melpomene 相应的同域种族之间共享遗传变异。
然而,并非基因组的所有部分都会受到同样的影响。特别是,我们怀疑 18 号染色体上的翅膀图案基因 optix 受到了强烈的选择。 optix 的差异调节可以导致翅膀上红色色素沉着的不同分布,如在 H. melpomene 的不同亚种中所见,或者导致红色色素缺失,如在 H. cydno 中所见。
赫利科尼乌斯翅膀图案可以警告捕食者它们有毒。有些物种参与缪勒拟态,有毒物种进化得彼此相似,这有助于加强捕食者的学习。模仿可以通过相同翅膀图案上的独立收敛来实现,也可以通过适应性基因渗入来交换翅膀图案等位基因来实现。因此,我们预测不同物种的共模仿种群可能在 optix 附近显示出过多的基因渗入信号。
我们对具有不同翅膀图案的种群有着完全不同的期望。如果某个地区的捕食者认为最常见的本地图案有毒,那么拥有外来翅膀图案的代价将会很高。同样,任何具有中间翅膀图案的混合个体也将面临更高的捕食风险。因此,我们预测,在具有不同翅型的种群之间,optix附近的基因渗入程度应该会减少。
量化整个基因组
简而言之,该检验使用三个群体和一个具有关系 (((P1,P2),P3),O) 的外群体,并调查 P2 和 P3 之间是否存在过多的共享变异(与 P1 和 P3 之间共享的变异相比) )。
这种过量可以用 D 统计量来表示,其范围从 -1 到 1,并且在无渗入的原假设下应等于 0。 D > 1 表示 P3 和 P2 之间可能存在基因渗入(或其他可能导致偏离严格的分叉物种历史的因素)。
该测试旨在用于全基因组规模。 D 统计量不太适合比较整个基因组的混合水平,因为它的绝对值取决于诸如有效种群大小等因素,而有效种群大小可能在整个基因组中有所不同。
其他教程中描述的 f 估计更好,因为它根据定义反映了混合比例,但它对小规模的随机误差高度敏感。因此,为此目的开发了一种称为 fd 的统计数据,该统计数据对于使用少量 SNP 引入的误差更加稳健。传统的 f 估计假设 P3 是供体群体,P2 是受者群体,而 fd 则在逐个地点的基础上推断供体。
选择人群
这些统计数据的解释很大程度上取决于所选人群。首先,该测试对从 P3 渗入 P2 最为敏感,而不是相反。
其次,fd 应被解释为 P3 和 P2 之间不与 P1 共享的过度共享变异的量化。如果 P1 和 P2 之间存在持续的基因流,则任何从 P3 到 P2 的基因渗入都会被低估。最后,我们只能量化比 P1 和 P2 之间的分裂更晚发生的渗入。
因此,如果我们想要量化整个基因组中发生的最大可检测量渗入,我们应该选择异域且与 P2 关系不太密切的 P1。不过,我们也可以通过测试这个特性来发挥我们的优势。如果我们选择与 P2 共享正在进行的基因流的 P1,那么测试将显示 P2 和 P3 共享 P1 不共享的基因组部分。这对于识别翅膀图案等位基因非常有用,因为这些通常是亚种在基因流中保持独特的唯一基因组区域。
实战
准备
打开终端窗口并导航到将运行练习并存储所有输入和输出数据文件的文件夹。现在创建一个名为“data”的子目录并下载本教程所需的数据文件
mkdir data
cd data
wget https://github.com/simonhmartin/tutorials/raw/master/ABBA_BABA_windows/data/hel92.DP8HET75MP9BIminVar2.chr18.geno.gz
wget https://github.com/simonhmartin/tutorials/raw/master/ABBA_BABA_windows/data/hel92.pop.txt
wget https://github.com/simonhmartin/tutorials/raw/master/ABBA_BABA_windows/data/chr18.LDhelmet_MLrho.w100.tsv
cd ..
- 接下来,在 GitHub 上下载本教程所需的 python 脚本集合
wget https://github.com/simonhmartin/genomics_general/archive/master.zip
unzip master.zip
滑动窗口分析
- 针对两个不同的情况运行分析 python 脚本。在这两种情况下,P1 都是全域性 H. melpomene melpomene (mel_mel)。 P2 和 P3 是我们期望共享基因的两个种群。在第一种情况下,我们量化了来自巴拿马的 H. melpomene rosina (mel_ros) 和 H. cydno chioneus (cyd_chi) 之间的基因渗入。在第二个例子中,我们量化了来自秘鲁的 H. melpomene amaryllis (mel_ama) 和 H. timareta thelxinoe (tim_txn) 之间的基因渗入。
python genomics_general-master/ABBABABAwindows.py \
-g data/hel92.DP8HET75MP9BIminVar2.chr18.geno.gz -f phased \
-o data/hel92.DP8HET75MP9BIminVar2.chr18.ABBABABA_mel_ros_chi_num.w25m250.csv.gz \
-P1 mel_mel -P2 mel_ros -P3 cyd_chi -O num \
--popsFile data/hel92.pop.txt -w 25000 -m 250 --T 2
python genomics_general-master/ABBABABAwindows.py \
-g data/hel92.DP8HET75MP9BIminVar2.chr18.geno.gz -f phased \
-o data/hel92.DP8HET75MP9BIminVar2.chr18.ABBABABA_mel_ama_txn_num.w25m250.csv.gz \
-P1 mel_mel -P2 mel_ama -P3 tim_txn -O num \
--popsFile data/hel92.pop.txt -w 25000 -m 250 --T 2
我们为脚本提供一个包含基因型数据 (-g) 的输入文件、一个输出文件 (-o)、内群体群体和外群体群体(-P1、-P2、-P3 和 -O)以及一个指定每个群体的文件示例位于(--popsFile)中。
我们还给出了窗口的参数。这些 ae“坐标”窗口,这意味着每个窗口相对于参考基因组的长度相同,但每个窗口的 SNP 数量可能不同。窗口大小 (-w) 将为 25,000 bp。 Windows 需要包含至少 (-m) 250 个 SNP 才能被视为有效。
最后,我们告诉脚本使用两个线程 (-T)。如果你有一个多核机器,你可以增加这个值,脚本会运行得更快。
我们需要将每个窗口统计文件加载到 R 中。我们将创建一个包含两个数据集的列表。首先输入输入文件的名称
AB_files <- c("data/hel92.DP8HET75MP9BIminVar2.chr18.ABBABABA_mel_ros_chi_num.w25m250.csv.gz",
"data/hel92.DP8HET75MP9BIminVar2.chr18.ABBABABA_mel_ama_txn_num.w25m250.csv.gz")
AB_tables = lapply(AB_files, read.csv)
head(AB_tables[[1]])
当 D 为负数时,fd 毫无意义,因为它旨在仅在存在过量时量化 ABBA 相对于 BABA 的过量。因此,我们在 D 为负数的位置将所有 fd 值转换为 0。
for (x in 1:length(AB_tables)){
AB_tables[[x]]$fd = ifelse(AB_tables[[x]]$D < 0, 0, AB_tables[[x]]$fd)
}
然后,我们可以为我们分析的两种情况绘制染色体上的 fd 图。
par(mfrow=c(length(AB_tables), 1), mar = c(4,4,1,1))
for (x in 1:length(AB_tables)){
plot(AB_tables[[x]]$mid, AB_tables[[x]]$fd,
type = "l", xlim=c(0,17e6),ylim=c(0,1),ylab="Admixture Proportion",xlab="Position")
rect(1000000,0,1250000,1, col = rgb(0,0,0,0.2), border=NA)
}
这表明染色体渗入程度存在相当大的异质性。如果我们考虑 optix 周围的区域,我们就会看到 H. melpomene rosina 和 H. cydno chioneus 之间基因渗入减少的证据,正如我们预测的那样。相比之下,我们看到了 H. melpomene amaryllis 和 H. timareta thelxinoe 之间基因渗入程度升高的证据,这表明它们共享的翅膀模式可能是适应性基因渗入的结果。鉴于这一证据,建议对 optix 周围区域进行系统发育,以测试 H. timareta 等位基因是否似乎“嵌套”在 H. melpomene 进化枝内。
基因渗入和重组率之间的关联
理论预测,如果存在许多选择基因渗入的“屏障位点”,我们应该会看到低重组区域基因渗入减少的趋势,因为中性基因渗入等位基因和有害基因之间的联系更强。我们可以通过检查重组率和整个染色体的 fd 之间的关系来检验这个假设。我们有一个先前生成的数据文件(已提供),给出了该染色体上 100 kb 窗口中估计的群体重组率。
现在我们将制作一个 100 kb 窗口的 fd 匹配数据集,这里仅使用显示渗入率最高的物种对:来自巴拿马的 H. melpomene rosina 和 H. cydno chioneus。
python ~/Research/genomics_general/ABBABABAwindows.py \
-g data/hel92.DP8HET75MP9BIminVar2.chr18.geno.gz -f phased \
-o data/hel92.DP8HET75MP9BIminVar2.chr18.ABBABABA_mel_ros_chi_num.w100m1.csv.gz \
-P1 mel_mel -P2 mel_ros -P3 cyd_chi -O num \
--popsFile data/hel92.pop.txt -w 100000 -m 1000 --T 2
现在,回到 R 中,读入这个新数据文件。
AB_table_w100 <- read.csv("data/hel92.DP8HET75MP9BIminVar2.chr18.ABBABABA_mel_ros_chi_num.w100m1.csv.gz")
和之前一样,我们将 D 为负的窗口的所有 fd 值转换为 0。
AB_table_w100$fd = ifelse(AB_table_w100$D < 0, 0, AB_table_w100$fd)
现在我们读取 100 kb 窗口的重组率表。这里,ML_rho 列给出了每个窗口的群体重组率的最大似然估计。
rec_table <- read.table("data/chr18.LDhelmet_MLrho.w100.tsv", header=T)
head(rec_table)
由于数据的噪声性质,我们想要比较不同重组率的 bin 中的 fd 值。我们将使用剪切函数将窗口分成三个具有低、中和高重组率的箱。
rec_bin <- cut(rec_table$ML_rho, 3)
最后,我们可以制作箱线图来比较这些箱之间推断的混合水平 (fd)。
boxplot(AB_table_w100$fd ~ rec_bin)
这证实了基因渗入水平确实随着重组率的增加而增加,这与大量屏障位点在全基因组范围内选择基因渗入的模型一致。








 苏公网安备32011502012024号
苏公网安备32011502012024号