1. 简介
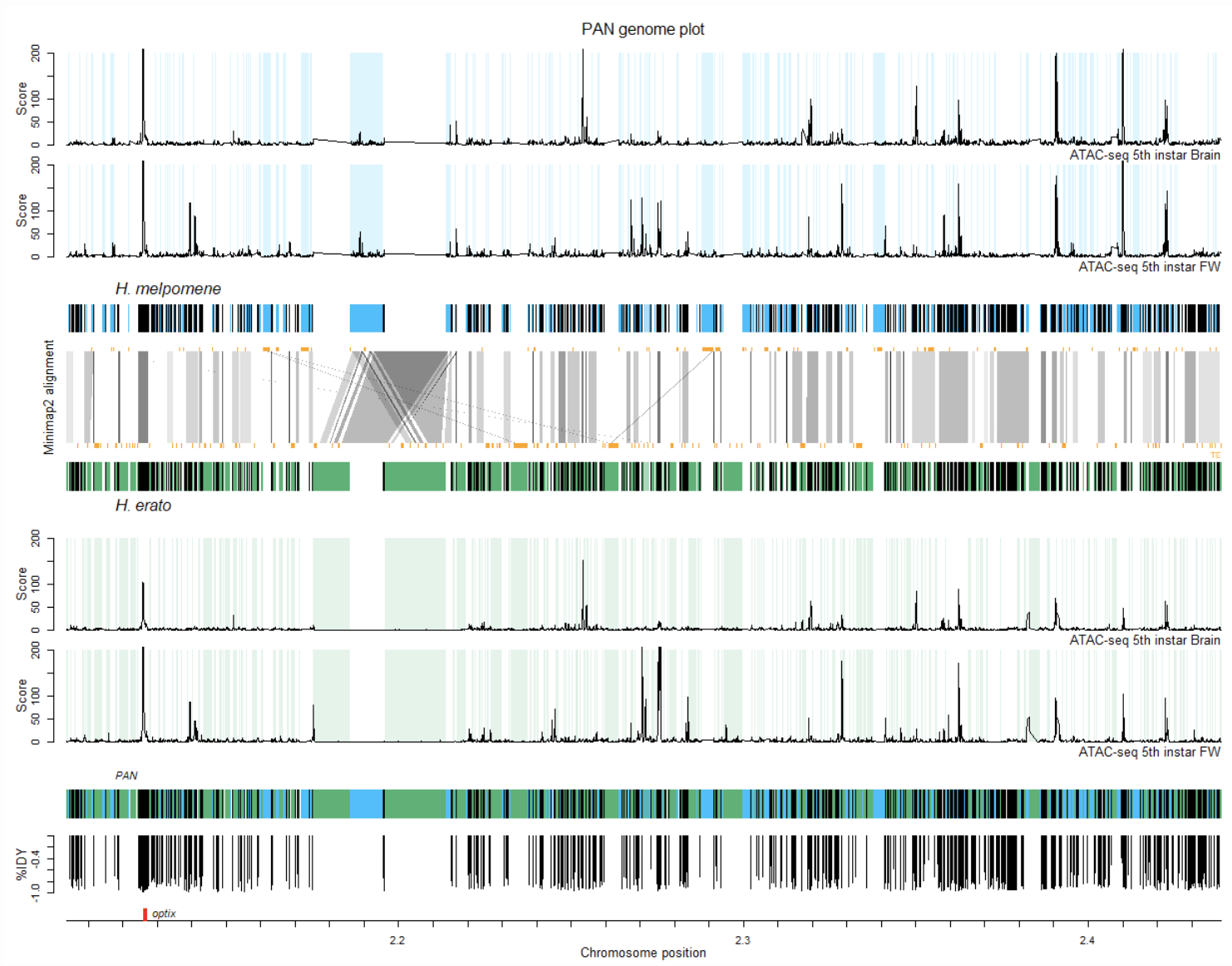
在本教程中,我们将把两个 Heliconius 蝴蝶物种的一条染色体(包含 optix 基因)与泛基因组进行比对。

泛基因组是指一个物种的所有个体共享的完整基因组序列,以及特定个体或亚群所独有的可变基因组序列。我们将使用 seq-seq-pan 构建泛基因组比对,使用一些自定义 Python 脚本来解析输出,并使用 R 来可视化比对。此外,我们将把发育中的头部和翅膀组织的转座元件(TE)注释和染色质可及性图谱(ATAC-seq)的坐标转换到泛基因组坐标空间,并将它们添加到该图中。
最终结果应如下所示:
2. 输入数据
对于本练习,您可以导航至 http://ensembl.lepbase.org/ 并单击 Heliconiuserato demophoon (v1) 和 Heliconius melpomene melpomene (Hmel2) 基因组组件的链接。在右上角,您可以搜索“optix”。搜索结果将返回基因模型名称(例如 evm.TU.Herato1801.64)及其位置(例如支架“Herato1801”位于“1239943”到“1251211”位置)。
# optix H. erato
Herato1801:1239943-1251211
# optix H. melpomene
Hmel218003:705604-706407
当你点击基因模型时,你可以看到 optix 基因周围有哪些基因。您还可以在左侧看到“导出数据”按钮。这允许您将序列导出为 .fasta 文件。使用此功能,您不仅可以尝试导出 optix 基因,还可以导出它周围的 2,000,000 bp 区域。
您还可以在此处找到这些 .fasta 文件。
# scaffold Herato1801 start 1 end 2000000
sequences/Herato1801_1_2000000.fasta
# scaffold Hmel213003 start 1 end 2000000
sequences/Hmel213003_1_2000000.fasta
3. seq-seq-pan aligner
我们可以使用 seq-seq-pan 将 fasta 文件中的序列组装成 pan 基因组。 Seq-seq-pan 通过构建复合共有序列或泛基因组来扩展多基因组比对器渐进式 Mauve 的功能,其中包括同源序列或局部共线块 (LCB) 以及每个基因组中的谱系特异性(非同源)序列基因组。然后将该泛基因组用作多基因组比对的参考坐标空间,其中包括任何基因组特有的序列。
将 fasta 文件中的序列组装成 pan 基因组。 Seq-seq-pan 通过构建复合共有序列或泛基因组来扩展多基因组比对器渐进式 Mauve 的功能,其中包括同源序列或局部共线块 (LCB) 以及每个基因组中的谱系特异性(非同源)序列基因组。然后将该泛基因组用作多基因组比对的参考坐标空间,其中包括任何基因组特有的序列。
对于我们的序列,我们将使用 seq-seq-pan,如下所示:
seq-seq-pan-wga --config genomefile=genome_list.txt outfilename=seq-seq-pan_out/SeqSeqPan_erato_melp_optix
Genome_list.txt 文件包含要包含在泛基因组组装中的 fasta 序列列表(每行一个)。该文件可以在这里下载。
Seq-seq-pan 将输出几个文件。其中有两个与我们相关:
- _consensus.fasta 文件包含共有泛基因组的完整 fasta 序列(将所有非同源序列拼接到组件中,并采用多个比对基因组中最常见的等位基因)。该共识文件划分了泛基因组的坐标空间,当我们想要将原始基因组中的任何位置(例如TE位置)映射到泛基因组时将使用该共识文件。
- .xmfa 文件包含局部共线块 (LCB) 的列表。我们将使用此文件来识别同源或物种特异性的序列。
- 1:genome_list.txt 文件中第一个基因组的序列标识符。
- 2:genome_list.txt 文件中第二个基因组的序列标识符。
= 区分单独的 LCB。- 对齐的 LCB 中存在间隙。
就是这样,我们有了泛基因组!
4. 共享和独特的序列
我们现在可以尝试确定序列的哪些部分在泛基因组中被识别为同源或物种特异性。为此,我们将使用自定义 Python 脚本,可在此处获取。
# For the Python script to work, we first need to remove newlines (enters) in the file from lines that include the sequence. This can be done with this perl oneliner:
perl -pe 'chomp if /^[ATCGNSBDHVMRWYK-]/' seq-seq-pan_out/SeqSeqPan_erato_melp_optix.xmfa| sed 's/\=/\n\=/g' | sed 's/>/\n>/g' | sed '/^$/d' > seq-seq-pan_out/SeqSeqPan_erato_melp_optix.noNewline.xmfa
# Now we can run the python script ('-g 1,2' is a list of the genome identifiers in the order of the genomes_list.txt file):
python seq-seq-pan_blocks_intervals.py -I seq-seq-pan_out/SeqSeqPan_erato_melp_optix.noNewline.xmfa -g 1,2
# The script annoyingly produces a 'TAB' at the end of each line which would break bedtools in the next step. We can remove that as follows:
sed -i 's/[[:space:]]*$//' seq-seq-pan_out/1_blocks_intervals.bed
sed -i 's/[[:space:]]*$//' seq-seq-pan_out/2_blocks_intervals.bed
输出是带有 | 的文件染色体|开始 |结束 |每个基因组中序列的位置,但在泛基因组的坐标空间中(因此,当该序列被另一个基因组中的物种特异性序列打断时,会生成一条新线)。这种具有开始和结束位置的格式通常称为 .bed 文件格式。
接下来,我们可以使用bedtools 这些文件,得到每个基因组中的独特序列。
bedtools subtract -sorted -a 1_blocks_intervals.bed -b 2_blocks_intervals.bed > blocks_unique_1.bed
bedtools subtract -sorted -b 1_blocks_intervals.bed -a 2_blocks_intervals.bed > blocks_unique_2.bed
我们还有一个自定义 Python 脚本来计算 LCB 内的序列同一性,可在此处获取。
# First, we need to transform the .xmfa file to a .fasta alignment
python seq-seq-pan_toFasta.py -I seq-seq-pan_out/SeqSeqPan_erato_melp_optix.noNewline.xmfa -g 1,2
cat genome* > seq-seq-pan_out/SeqSeqPan_erato_melp_optix.fasta
# Next, we need to find the sequences that are shared between genomes. We can do this with bedtools intersect.
bedtools intersect -sorted -a seq-seq-pan_out/1_blocks_intervals.bed -b seq-seq-pan_out/2_blocks_intervals.bed > seq-seq-pan_out/blocks_shared_1_2.bed
# Finally, we can calculate the conservation. This will output a .bed like file with identity scores for each shared interval.
python seq-seq-pan_bedfile_conservation.py -I seq-seq-pan_out/SeqSeqPan_erato_melp_optix.fasta -g 1,2 -b seq-seq-pan_out/blocks_shared_1_2.bed -o seq-seq-pan_out/conservation_shared_1_2.bed
5. 将注释映射到泛基因组
seq-seq-pan 的映射功能允许将所包含基因组的任何原始位置转换为泛基因组(=泛基因组坐标)。该函数将一个文件作为输入,该文件包含单列位置和第一行,该文件指定从何处映射到何处(例如 2\tc,这意味着从基因组 2 进行映射(Hmel218003 序列,它是基因组列表中的第二个基因组) .txt 文件)到基因组 c(共识泛基因组序列))。
这里我有 Hmel218003 支架上 optix 基因的起始和结束位置(您也可以从 http://ensembl.lepbase.org/ 获取这些或任何基因的位置。
2 c
705604
706407
运行映射函数:
seq-seq-pan map -c seq-seq-pan_out/SeqSeqPan_erato_melp_optix_consensus.fasta -p ./ -i gene_annotation/optix_Hmel2.toMap.txt -n gene_annotation/optix_Hmel2.toMap.pan
现在让我们在 R 中绘制泛基因组。








 苏公网安备32011502012024号
苏公网安备32011502012024号