导读
本文将介绍基因组组装过程中,如何利用HiC测序数据,进行染色体级别基因组的组装。该过程主要利用 Juicer 和 3D-DNA 进行,有关第一步Juicer的过程,已经下方的文章中介绍了,本文主要介绍第二步:3D-DNA的安装与使用。
1. 背景介绍
目前基因组组装的主要流程是,利用二代或者三代技术进行测序,利用得到的测序数据,拼接为contig级别的基因组,如果需要上升到染色体级别,那么就需要对物种进行HiC测序,进一步进行染色体挂载。目前对于二倍体动物,3D-DNA是效果最好的,下面就介绍3D-DNA的使用方法。
2. 安装
2.1. 流程图
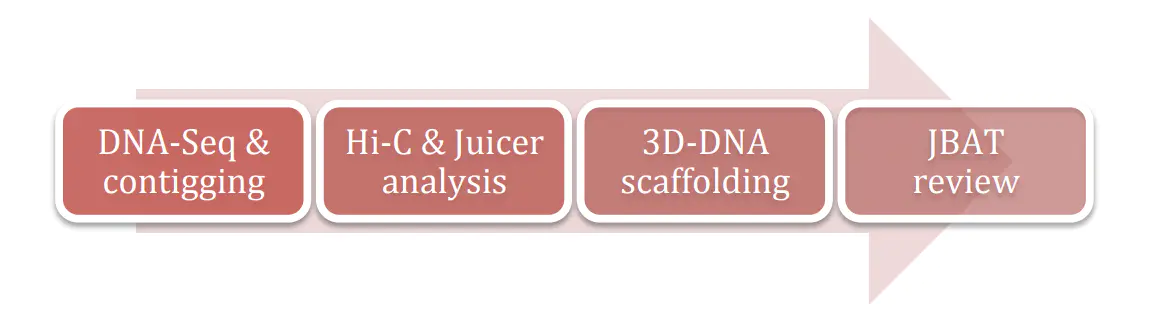
上图是使用3D-DNA进行染色体挂载的流程图,其中第一步是测序和基础组装,测序一般是交给测序公司来完成,contig组装利用物种对应的组装软件即可。第二步时利用Juicer对HiC数据进行分析。第三步是利用3D-DNA进行挂载。
2.2. 依赖
下面这些是3D-DNA的依赖,如果阅读过之前Juicer的使用教程,那么环境已经配置完成,没有阅读过的读者,可以在文末选择跳转。
LastZ (version 1.03.73 released 20150708) –仅适用于二倍体Java version >=1.7Bash >=4GNU Awk >=4.0.2GNU coreutils sort >=8.11Python >=2.7 - 仅适用于染色体编号感知分离器模块scipy numpy matplotlib - 仅适用于染色体编号感知分离器模块
2.3. clone
# 从Github拉取仓库
git clone https://github.com/theaidenlab/3d-dna.git
3. 实战
3.1. 数据准备
- 基因组文件:
genome.fa
Juicer结果:merged_nodups.txt
3.2. run
# 对组装的信心高,用-r 0, 否则用默认的-r 2就行了
# -r 代表 3d-dna 修正的次数
# merged_nodups.txt 在 上一步Juicer运行的aligned目录下
/home/ubuntu/3d-dna/run-asm-pipeline.sh -r 2 \
reference/genome.fa aligned/merged_nodups.txt &> log.txt &
3.3. 结果
最终的输出文件最关键的是下面三类:
.fasta: 以FINAL标记的是最终结果.hic: 各个阶段都会有输出结果,用于在JABT中展示.assembly: 各个阶段都会有输出,一共两列,存放contig的组装顺序
将结果中的.hic文件和.assembly文件导入Juicebox中进行调整,最后输出修改后的.assembly文件,再运行下面命令,即可获取染色体级别的基因组。
/home/ubuntu/3d-dna/run-asm-pipeline-post-review.sh \
-r genome.review.assembly \
genome.fa aligned/merged_nodups.txt
# genome.review.assembly 来自Juicebox中导出
读者有任何问题,可在评论区进行交流。
| 







 苏公网安备32011502012024号
苏公网安备32011502012024号