1. 寻找开发区域
ATACseq 的一个共同目标是识别转录因子结合和/或转录机制活跃的无核小体区域。该核小体游离信号对应于小于一个核小体的片段(如 Greenleaf 论文中定义 < 100bp)。
然而,为了识别开放的染色质,我们可以简单地使用在测序中正确配对的所有读数(< 2000bp)。
2. 无核小体区域
有许多方法可用于从 ATACseq 数据中调用无核小体区域,其中许多方法借鉴自 ChIPseq 分析。一种非常流行且标准的 ATACseq 峰值调用程序是 MAC2。
2.1. 单端数据
使用 ATACseq 的单端测序,我们不知道片段有多长。因此,与 ChIPseq 相比,要识别开放区域,MACS2 峰值调用需要一些不同的参数。采用的一种策略是将读取 5' 末端移动 -100,然后从该位置延伸 200bp。考虑到我们的无核小体片段的预期大小,这应该提供适合 MACS2 窗口大小的核小体区域的堆积。
这是我们用来执行此操作的 MACS 系统调用。
MACS2 callpeak -t singleEnd.bam --nomodel --shift -100
--extsize 200 --format BAM -g MyGenome
在 R 中,我们可以使用 Herper 运行这个系统调用,这样我们就可以访问我们安装的 MACS2。 MACS2 已安装到 ATACseq_analysis 中。所以我们可以使用 with_CondaEnv() 从 R 中使用这个环境。
with_CondaEnv("ATACseq_analysis",
system2(command="macs2",args =c("callpeak",
"-t", "singleEnd.bam",
"--nomodel",
"--shift","-100",
"--extsize", "200",
"--format", "BAM",
"-g", "hs")),
stdout = TRUE))
或者对于核小体占据的数据,我们可以调整移位和延伸以将信号集中在核小体中心(包裹在 147bp DNA 中的核小体)。
MACS2 callpeak -t singleEnd.bam --nomodel --shift 37
--extsize 73 --format BAM -g MyGenome
with_CondaEnv("ATACseq_analysis", system2(command = "macs2", args = c("callpeak",
"-t", "singleEnd.bam", "--nomodel", "--shift", "37", "--extsize", "73", "--format",
"BAM", "-g", "hs")), stdout = TRUE)
2.2. 双端数据
如果我们对配对末端数据进行了测序,那么我们确实知道片段长度,并且可以向 MACS2 提供 BAM 文件,这些文件已经过预过滤以正确配对(如果您想区分核小体和无核小体区域,则提供片段大小)。
我们必须告诉 MACS2 数据是使用格式参数配对的。这很重要,因为默认情况下 MACS2 会猜测它是单端 BAM。
MACS2 callpeak -t pairedEnd.bam -f BAMPE
--outdir path/to/output/
--name pairedEndPeakName -g MyGenome
with_CondaEnv("ATACseq_analysis", system2(command = "macs2", args = c("callpeak",
"-t", "pairedEnd.bam", "--format", "BAMPE", "--outdir", "/Documents/ATAC_MACS2_calls/",
"--name", "pairedEndPeakName", "-g", "hs")), stdout = TRUE)
对于此处的配对末端数据,我们从无核小体区域 BAM 文件中调用无核小体区域的峰。
MACS2 callpeak -t ~/Downloads/Sorted_ATAC_50K_2_openRegions.bam
--outdir ATAC_Data/ATAC_Peaks/ATAC_50K_2
--name Sorted_ATAC_50K_2_Small_Paired_peaks.narrowPeak
-f BAMPE -g hs
with_CondaEnv("ATACseq_analysis", system2(command = "macs2", args = c("callpeak",
"-t", "~/Downloads/Sorted_ATAC_50K_2_openRegions.bam", "--outdir", "ATAC_Data/ATAC_Peaks/ATAC_50K_2",
"--name", "Sorted_ATAC_50K_2_Small_Paired_peaks.narrowPeak", "-f", "BAMPE", "-g",
"hs")), stdout = TRUE)
在之后,我们将获得 3 个文件。
- Name.narrowPeak – 适用于 IGV 和进一步分析的格式
- Name_peaks.xls – 适合在 excel 中查看的峰值表。(实际上不是 xls,而是 tsv)
- summits.bed – 用于查找
motifs 和绘图的峰顶位置
3. QC
通常我们想做一些 QC 来检查低质量、重复和信号分布。在我们删除任何数据之前,我们可以快速评估我们的峰值读取、重复率、低质量读取和来自 ChIPQC 的伪像区域中的读取。
library(ChIPQC)
library(rtracklayer)
library(DT)
library(dplyr)
library(tidyr)
blkList <- import.bed("~/Downloads/ENCFF001TDO.bed.gz")
openRegionPeaks <- "~/Downloads/ATAC_Workshop/ATAC_Data/ATAC_Peaks/Sorted_ATAC_50K_2_Small_Paired_peaks.narrowPeak"
qcRes <- ChIPQCsample("~/Downloads/ATAC_Workshop/ATAC_Data/ATAC_BAM/Sorted_ATAC_50K_2_openRegions.bam",
peaks = openRegionPeaks, annotation = "hg19", chromosomes = "chr20", blacklist = blkList,
verboseT = FALSE)
我们可以使用 ChIPQC 包来捕获我们的 ATACseq 数据的一些重要指标,例如来自 QCmetrics() 函数的峰值读取和黑名单中的读取以及来自 flagtagcounts() 函数的重复读取数。
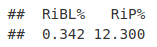
myMetrics <- QCmetrics(qcRes)
myMetrics[c("RiBL%", "RiP%")]
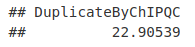
flgCounts <- flagtagcounts(qcRes)
DupRate <- flgCounts["DuplicateByChIPQC"]/flgCounts["Mapped"]
DupRate * 100
4. 黑名单删除
来自测序的人工制品和不完美的基因组构建可能会混淆我们的结果。这些工件已被整理到区域的“黑名单”中。
由于列入黑名单的区域可能会混淆我们的分析,因此我们删除了在那里被调用的所有峰值。
过早删除黑名单可能会隐藏数据中的一些 QC 问题。在您的分析中应始终考虑黑名单,并建议在考虑 QC 后从这些区域中删除数据。
MacsCalls <- granges(qcRes[seqnames(qcRes) %in% "chr20"])
data.frame(Blacklisted = sum(MacsCalls %over% blkList), Not_Blacklisted = sum(!MacsCalls %over%
blkList))
MacsCalls <- MacsCalls[!MacsCalls %over% blkList]








 苏公网安备32011502012024号
苏公网安备32011502012024号