ATACseq
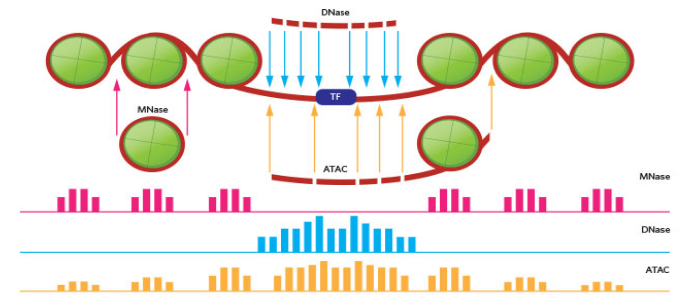
ATACseq - 使用转座酶并提供一种同时从单个样本的转录因子结合位点和核小体位置提取信号的方法。
1. 数据类型
上面这意味着我们的数据中可能包含多种信号类型。
- 我们将从无核小体区域和转录因子(我们的较短片段)周围获得信号。
- 我们的一部分信号将来自开放染色质(较长片段)中的核小体周围。
我们所有的数据都来自我们的转座酶能够访问的开放染色质。
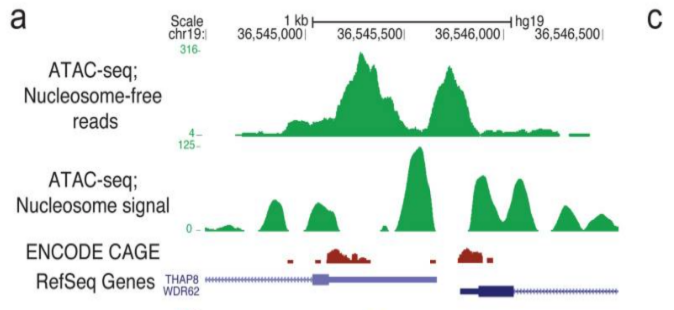
2. 评估 TSS 信号
2.1. TSS 区域
如果我们的较短片段代表转录因子和转录机制周围的开放区域,我们希望在转录起始位点看到信号。
我们较长的片段将代表核小体周围的信号,因此信号应该在转录起始位点之外,更多地出现在 +1 和 -1 核小体位置。
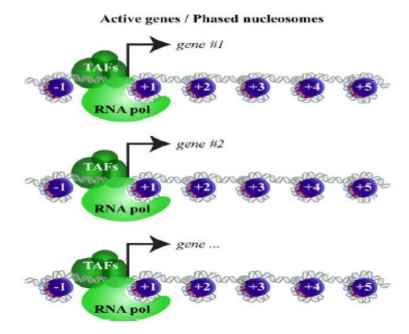
我们可以在所有 TSS 区域创建一个图,以说明我们的核小体游离和核小体占据的信号部分最普遍的位置。Meta-plots 在区域集上平均或求和信号以识别数据趋势。
2.2 可视化
要生成区域信号的图,我们可以使用 soGGi bioconductor 包。我们可以使用 BiocManager::install 和库函数加载 soGGi。
BiocManager::install("soGGi")
library(soGGi)
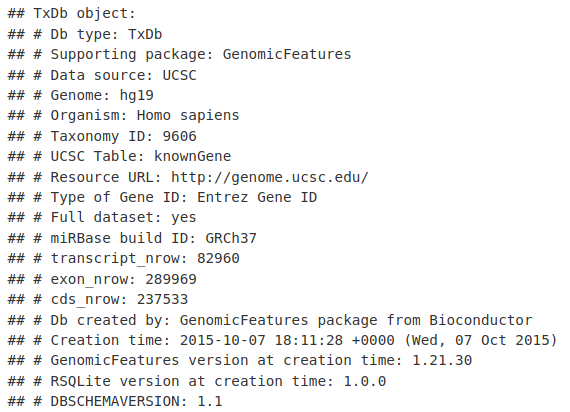
soGGi 库只需要一个 BAM 文件和一个 GRanges 区域,在这些区域上平均信号以生成图。我们希望绘制 TSS 区域,因此我们首先需要为 hg19 基因组生成 TSS 位置的 GRanges。首先,我们可以加载我们感兴趣的 TxDb - TxDb.Hsapiens.UCSC.hg19.knownGene。
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
TxDb.Hsapiens.UCSC.hg19.knownGene
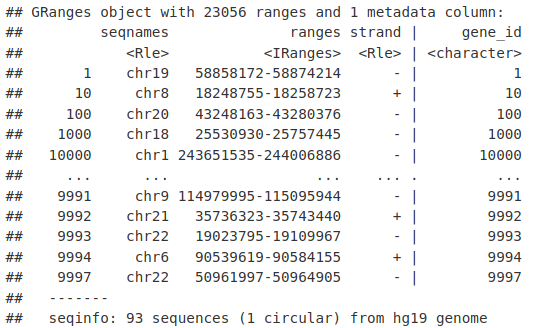
我们可以使用 genes() 函数和我们的 TxDb 对象提取基因位置(TSS 到 TTS)。
genesLocations <- genes(TxDb.Hsapiens.UCSC.hg19.knownGene)

genesLocations
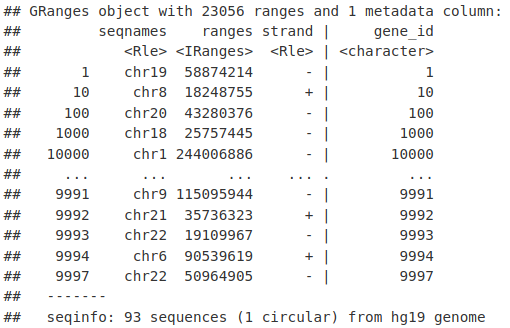
然后我们可以使用 resize() 函数提取每个基因(TSS)的起始位置。这里我们将固定位置设置为开始,宽度设置为 1。
tssLocations <- resize(genesLocations, fix = "start", width = 1)
tssLocations
当我们创建索引时,我们将基因组子集化为主要染色体。我们可以使用 TSS GRange 对象再次执行此操作,并更新级别。这意味着 BAM 和 GRanges 会很好地发挥作用。
mainChromosomes <- paste0("chr", c(1:21, "X", "Y", "M"))
myindex <- (match(seqnames(tssLocations), mainChromosomes))
tssLocations <- tssLocations[as.numeric(myindex)]
seqlevels(tssLocations) <- mainChromosomes
soGGi 包的 regionPlot() 函数需要一个 BAM 数据文件来绘制提供给 bamFile 参数和一个 GRanges 来绘制提供给 testRanges 参数。
library(soGGi)
sortedBAM <- "~/Downloads/ATAC_Workshop/Sorted_ATAC_50K_2.bam"
library(Rsamtools)
# Nucleosome free
allSignal <- regionPlot(bamFile = sortedBAM, testRanges = tssLocations)
一个有用的功能是我们可以使用 minFragmentLength 和 maxFragmentLength 参数指定要在我们的绘图中使用的配对读取的最小和最大片段长度。这使我们能够仅选择我们的核小体自由信号(< 100 个碱基对)来生成我们在 TSS 区域的图。
nucFree <- regionPlot(bamFile = sortedBAM, testRanges = tssLocations, style = "point",
format = "bam", paired = TRUE, minFragmentLength = 0, maxFragmentLength = 100,
forceFragment = 50)
class(nucFree)
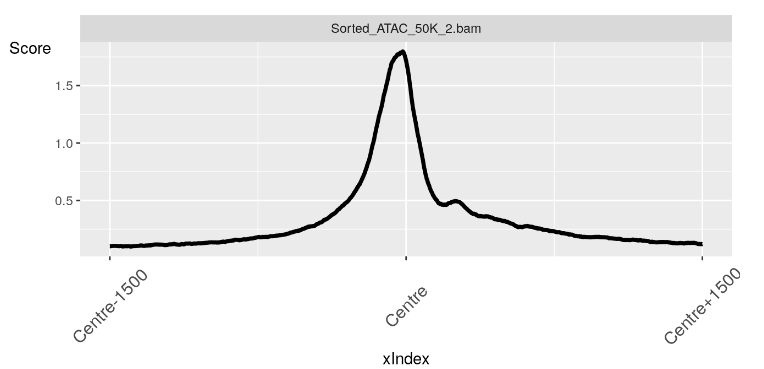
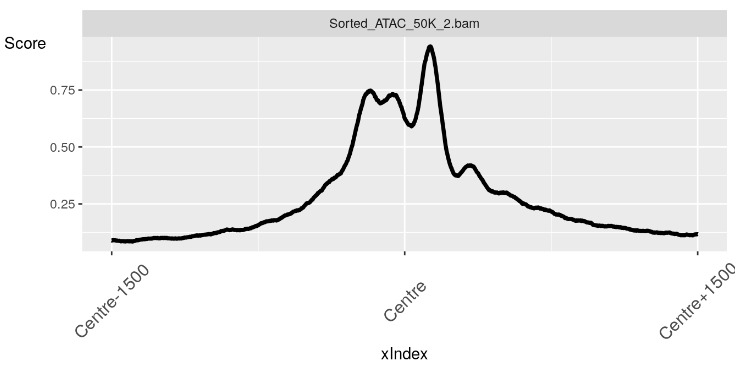
现在我们有了我们的配置文件对象,我们可以使用 soGGi 中的 plotRegion() 函数创建我们的图。
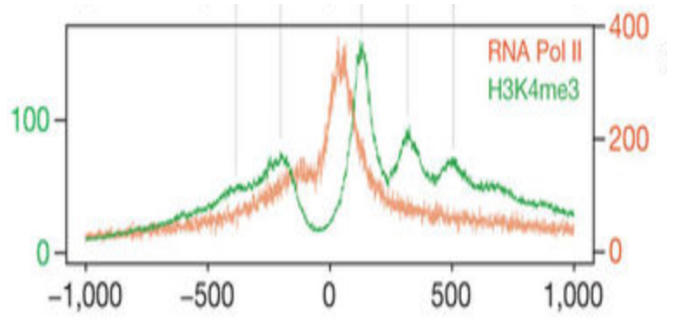
在这里,我们看到了 TSS 上方区域中无核小体区域的预期信号峰值。
plotRegion(nucFree)
我们可以通过将 minFragmentLength 和 maxFragmentLength 参数调整为核小体长度片段的预期参数(此处为 180 到 240)来为我们的单核小体信号创建一个图。
monoNuc <- regionPlot(bamFile = sortedBAM, testRanges = tssLocations, style = "point",
format = "bam", paired = TRUE, minFragmentLength = 180, maxFragmentLength = 240,
forceFragment = 80)
同样,我们可以使用 plotRegion() 函数在 TSS 位置绘制单核小体信号。在此图中,我们可以清楚地看到预期的 +1 核小体信号峰以及其他几个核小体信号峰。
plotRegion(monoNuc)








 苏公网安备32011502012024号
苏公网安备32011502012024号