学习内容
- 了解可用的基因组注释数据库和存储信息的不同类型
- 比较和对比可用于基因组注释数据库的工具
- 应用各种 R 包检索基因组注释
基因组注释
对二代测序结果的分析需要将基因、转录本、蛋白质等与功能或调控信息相关联。为了对基因列表进行功能分析,我们通常需要获得与我们希望使用的工具兼容的基因标识符。在这里,我们讨论了您可以获得基因注释信息的方法以及每种方法的一些优缺点。
数据库
我们从存储信息的必要数据库中检索有关过程、途径等(涉及基因的信息)的信息。您选择的数据库将取决于您要获取的信息类型。经常查询的数据库示例包括:
通用数据库
提供有关基因组特征、坐标、同源性、变异信息、表型、蛋白质域/家族信息、相关生物过程/途径、相关 microRNA 等的综合信息:
- Ensembl (use Ensembl gene IDs)
- NCBI (use Entrez gene IDs)
- UCSC
- EMBL-EBI
特定数据库
提供与特定主题相关的注释:
- Gene Ontology (GO): 基因本体生物过程、细胞成分和分子功能数据库——基于 Ensembl 或 Entrez 基因 ID 或官方基因符号
- KEGG: 生物通路数据库——基于 Entrez 基因 ID
- MSigDB: database of gene sets
- Reactome:生物通路数据库
- Human Phenotype Ontology: 与人类疾病相关的基因数据库
- CORUM: 人、小鼠、大鼠的蛋白质复合物数据库
- …
这不是一个详尽的列表,还有许多其他可用的数据库未在此处列出。
基因组
在开始搜索任何这些数据库之前,您应该知道使用了哪个基因组来生成您的基因列表,并确保在功能分析期间使用相同的进行注释。当获得新的基因组时,基因组特征(基因、转录本、外显子等)的名称和/或坐标位置可能会发生变化。因此,关于基因组特征(基因、转录本、外显子等)的注释是特定于基因组构建的,我们需要确保我们的注释是从适当的资源中获得的。
例如,如果我们使用人类基因组的 GRCh38 来量化用于差异表达分析的基因表达,那么我们应该使用相同的基因组 GRCh38 来在基因 ID 之间转换并识别每个基因的注释。
注释工具
在 R 中,有许多流行的包用于基因/转录本级别的注释。这些软件包提供的工具可以获取您提供的基因列表,并使用上面列出的一个或多个数据库检索每个基因的信息。
注释工具:用于访问/查询来自特定数据库的注释
| 工具 |
描述 |
优点 |
缺点 |
| org.Xx.eg.db |
查询目标生物的基因特征信息 |
基因ID转换、生物型和坐标信息 |
只有最新的基因组可用 |
| EnsDb.Xx.vxx |
直接从 Ensembl API 获取的转录本和基因级信息(类似于 TxDb,但具有过滤能力并由 Ensembl 版本进行版本控制) |
易于提取特征,直接过滤 |
不是最新的注解,比一些包更难用 |
| TxDb.Xx.UCSC.hgxx.knownGene |
用于转录本和基因水平信息的 UCSC 数据库,或者可以使用 GenomicFeatures 包从 SQLite 数据库文件创建自己的 TxDb |
特征信息,简单函数提取特征 |
只有当前和最近的基因组可用——可以创建你自己的 |
| annotables |
可用于人类和模式生物的基因级特征信息 |
超级快速和简单的基因 ID 转换、生物型和坐标信息 |
静态资源,不定期更新 |
| biomaRt |
Ensembl BioMart 在线工具的 R 包版本 |
所有可用的 Ensembl 数据库信息,Ensembl 上的所有生物,信息丰富 |
|
查询工具
接口工具:用于访问/查询来自多个不同注释源的注释
AnnotationDbi:查询 OrgDb、TxDb、Go.db、EnsDb 和 BioMart 注释。AnnotationHub:查询大量全基因组资源,包括ENSEMBL、UCSC、ENCODE、Broad Institute、KEGG、NIH Pathway Interaction Database等。
AnnotationDbi
AnnotationDbi 是一个 R 包,它提供了一个接口,用于连接和查询使用 SQLite 数据存储的各种注释数据库。AnnotationDbi 包可以查询 OrgDb、TxDb、EnsDb、Go.db 和 BioMart 注释。从这些数据库中提取数据时,可以参考文档。
虽然 AnnotationDbi 是一个流行的工具,但我们不会通过代码来使用这个包。但是,如果您对更多细节感兴趣,我们在此处提供了材料链接以及使用我们当前数据集的示例。
AnnotationHub
AnnotationHub 是访问基因组数据或查询大量全基因组资源的绝佳资源,包括 ENSEMBL、UCSC、ENCODE、Broad Institute、KEGG、NIH Pathway Interaction Database 等。所有这些信息都已存储并可通过直接连接轻松访问数据库。
要开始使用 AnnotationHub,我们首先加载库并连接到数据库:
# Load libraries
library(AnnotationHub)
library(ensembldb)
# Connect to AnnotationHub
ah <- AnnotationHub()
要查看存储在我们数据库中的信息类型,我们只需键入对象的名称:
# Explore the AnnotationHub object
ah
使用输出,您可以了解可以在 AnnotationHub 对象中查询的信息:
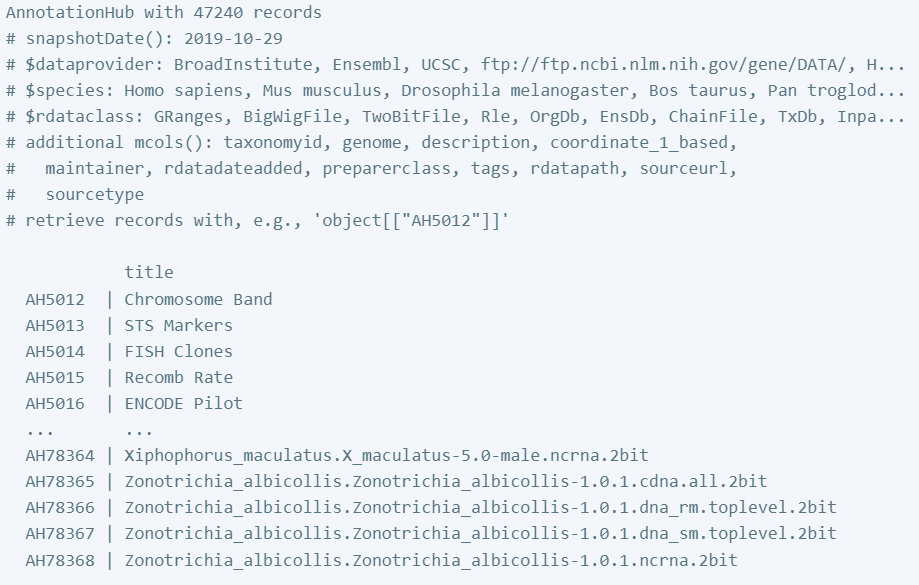
请注意有关使用对象 [[AH2]] 检索记录的注释 - 这将是我们如何从 AnnotationHub 对象中提取单个记录的方法。
如果您想查看有关任何数据类别的更多信息,您也可以提取该信息。例如,如果您想确定所有可用的物种信息,您可以在 AnnotationHub 对象中探索它:
# Explore all species information available
unique(ah$species) %>% View()
除了物种信息外,还有关于数据对象类型和数据提供者的附加信息:
# Explore the types of Data Objects available
unique(ah$rdataclass) %>% View()
# Explore the Data Providers
unique(ah$dataprovider) %>% View()
现在我们知道了 AnnotationHub 可用的信息类型,我们可以使用 query() 函数查询它以获得我们想要的信息。假设我们想返回人类的 Ensembl EnsDb 信息。要返回可用的记录,我们需要使用从 ah 对象输出的术语来提取所需的数据。
# Query AnnotationHub
human_ens <- query(ah, c("Homo sapiens", "EnsDb"))
查询检索 EnsDb 对象的所有匹配项,您将看到它们按版本号列出。 GRCh38 的最新版本是 Ensembl98,AnnotationHub 提供了它作为使用选项。但是,如果您查看旧版本的选项,对于智人,它只能追溯到 Ensembl 87。如果您使用的是 GRCh38,这很好,但是如果您使用的是像 hg19/GRCh37 这样的旧基因组构建,您将需要加载 EnsDb 包(如果该版本可用),或者您可能需要使用 ensembldb 构建自己的包。
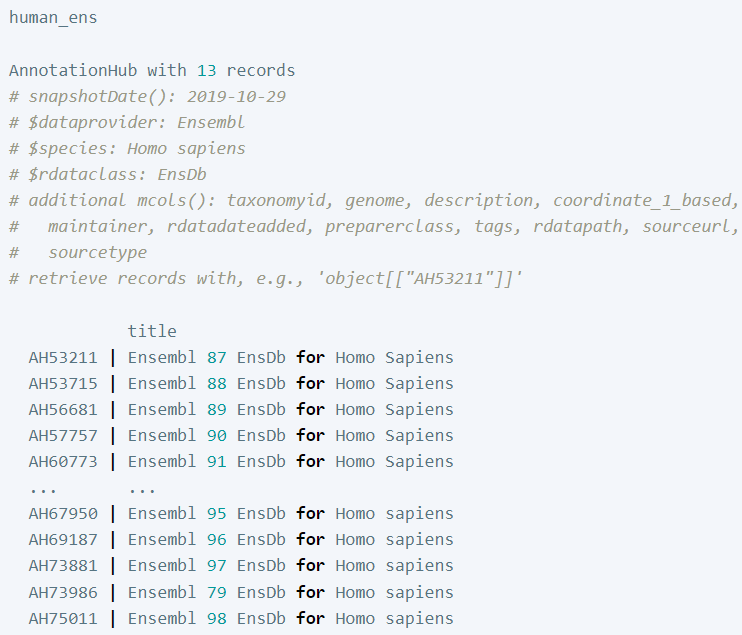
在我们的例子中,我们正在寻找最新的 Ensembl 版本,以便注释是最新的。要从 AnnotationHub 中提取此信息,我们可以使用 AnnotationHub ID 对对象进行子集化:
# Extract annotations of interest
human_ens <- human_ens[["AH75011"]]
现在我们可以使用 ensembldb 函数来提取基因、转录本或外显子级别的信息。我们对基因级注释感兴趣,因此我们可以按如下方式提取该信息:
# Extract gene-level information
genes(human_ens, return.type = "data.frame") %>% View()
但请注意,获取转录本或外显子级信息同样容易:
# Extract transcript-level information
transcripts(human_ens, return.type = "data.frame") %>% View()
# Extract exon-level information
exons(human_ens, return.type = "data.frame") %>% View()
要使用 AnnotationHub 获取注释数据框,我们将使用 genes() 函数,但只保留选定的列并过滤掉行,以保留与我们的基因标识符相对应的那些在我们的结果文件中:
# Create a gene-level dataframe
annotations_ahb <- genes(human_ens, return.type = "data.frame") %>%
dplyr::select(gene_id, gene_name, entrezid, gene_biotype) %>%
dplyr::filter(gene_id %in% res_tableOE_tb$gene)
这个 dataframe 看起来应该没问题,但是我们仔细看一下,我们会注意到包含 Entrez 标识符的列是一个列表,实际上有许多 Ensembl 标识符映射到多个 Entrez 标识符!
# Wait a second, we don't have one-to-one mappings!
class(annotations_ahb$entrezid)
which(map(annotations_ahb$entrezid, length) > 1)
让我们来看看我们的 Ensembl 标识符中有多少具有关联的基因符号,以及其中有多少是唯一的:
which(is.na(annotations_ahb$gene_name)) %>% length()
which(duplicated(annotations_ahb$gene_name)) %>% length()
让我们识别非重复的基因,只保留不重复的基因:
# Determine the indices for the non-duplicated genes
non_duplicates_idx <- which(duplicated(annotations_ahb$gene_name) == FALSE)
# How many rows does annotations_ahb have?
annotations_ahb %>% nrow()
# Return only the non-duplicated genes using indices
annotations_ahb <- annotations_ahb[non_duplicates_idx, ]
# How many rows are we left with after removing?
annotations_ahb %>% nrow()
最后,最好知道有多少 Ensembl 标识符映射到 Entrez 标识符:
# Determine how many of the Entrez column entries are NA
which(is.na(annotations_ahb$entrezid)) %>% length()
那是我们一半以上的基因!如果我们计划使用 Entrez ID 结果进行下游分析,我们一定要牢记这一点。如果您查看我们返回 NA 的查询中的一些 Ensembl ID,它们会映射到假基因(即 ENSG00000265439)或非编码 RNA(即 ENSG00000265425)。数据库之间的差异(我们可以预期观察到)是由于每个数据库都实现了自己不同的计算方法来生成基因构建。
- 使用
AnnotationHub 创建我们的 tx2gene 文件
要创建我们的 tx2gene 文件,我们需要结合使用上述方法并将两个数据帧合并在一起。例如:
# Create a transcript dataframe
txdb <- transcripts(human_ens, return.type = "data.frame") %>%
dplyr::select(tx_id, gene_id)
txdb <- txdb[grep("ENST", txdb$tx_id),]
# Create a gene-level dataframe
genedb <- genes(human_ens, return.type = "data.frame") %>%
dplyr::select(gene_id, gene_name)
# Merge the two dataframes together
annotations <- inner_join(txdb, genedb)
在本课中,我们的重点是使用注释包来提取信息,主要用于我们在下游使用的不同工具的基因 ID 转换。我们提供的许多注释包所包含的信息比我们进行功能分析所需的信息要多得多,我们在这里只是触及了皮毛。很高兴了解我们使用的工具的功能,因此我们鼓励您花一些时间探索这些包以更加熟悉它们。








 苏公网安备32011502012024号
苏公网安备32011502012024号