
一边学习,一边总结,一边分享!
本期教程
获得本教程 Data and Code,请在后台回复:20240811。
2022年教程总汇
2023年教程总汇
写在前面
我们在前面的教程分享了模式植物构建orgDb数据库 | 以org.Slycompersicum.eg.db为例,这两天也做对应的分析,但是发现前面的代码速度太慢了,以及会出现报错的情况。
那么我们也做了对应的优化,可以大大减少花费时间,以及减少我们的报错。
现在我们的代码,可以直接读取eggnog分析获得数据,非常的方便。
此外,我们在这里依旧推荐大家使用本地的 Egg-mapper进行注释,我自己使用下来,非常的快捷。虽然网页版的Egg-mapper工具,现在可以支持最多10000条的序列,但是速度依旧是比较慢的。
此外,本地使用Egg-mapper使用时,数据库的下载可能对新手小白具有挑战性,若是本推文阅读量超过5000+,我们也会视频讲解+文本,手把手教大家。
Code
本次,我们依旧使用我们的一步法进行数据库的构建。
source("../Set_OrgDb_Database.R")
# 使用函数
# 使用函数
Set_OrgDb_Database(
emapper_file = "tomato.emapper.annotations", ## 输入的eggnog结果文件
json_file = "../ko00001.json", ## 下载ko00001.json,下载网址:https://www.genome.jp/kegg-bin/get_htext?ko00001
tax_id = "4081", ## 物种信息
genus = "Solanum",
species = "lycompersicum",
versions = "4.0", ## 版本号
maintainer = "du<16***@qq.com>", ## 修改为你的名字和邮箱
author = "du<16****@qq.com>", ## 修改为你的名字和邮箱
outputDir = "."## 保存路径
)
我们自己这边注注释 番茄、黄瓜、甜瓜、辣椒的数据库,基本每个数据库会在2分钟内完成。
加载数据包并进行分析
install.packages("org.Slycompersicum.eg.db/", repos = NULL,
type = 'sources')
library(org.Slycompersicum.eg.db)
GO 富集分析
Gene_list <- read.table("01.tomato_target_gene.id.txt",header = F)$V1
head(Gene_list)
go_result <- enrichGO(gene = Gene_list,
OrgDb = org.Slycompersicum.eg.db,
keyType = "GID",
ont = "ALL",
qvalueCutoff = 1,
pvalueCutoff = 1)
go_data <- as.data.frame(go_result)
dim(go_data)
head(go_data)
write.csv(go_data, "02.tomato.miRNA.GO.result.csv")
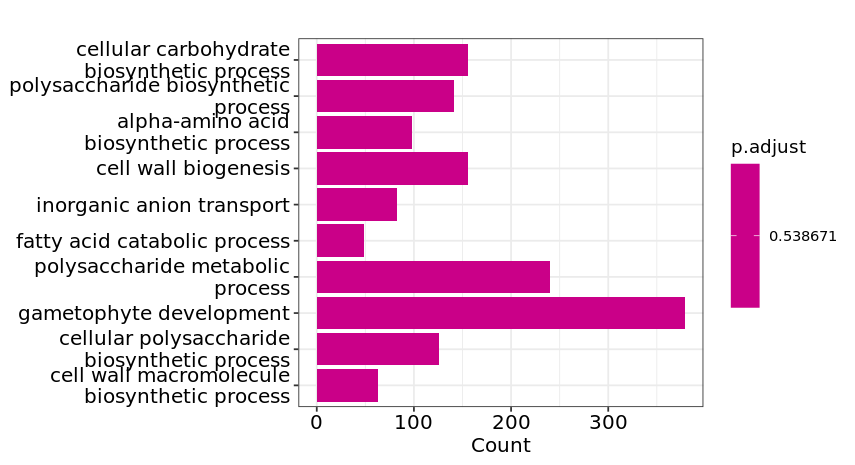
## 绘图
pdf("04.tomato.go.柱状图.pdf",width = 8, height = 10)
barplot(go_result,
drop = TRUE,
showCategory = 20)
dev.off()
KEGG 富集分析
##KEGG 富集分析
pathway2gene <- AnnotationDbi::select(org.Slycompersicum.eg.db,
keys = keys(org.Slycompersicum.eg.db),
columns = c("Pathway","Ko")) %>%
na.omit() %>%
dplyr::select(Pathway, GID)
必须加载的数据,在数据包制作过程中会生成。
load("kegg_info.RData")
kegg_result <- enricher(Gene_list,
TERM2GENE = pathway2gene,
TERM2NAME = pathway2name,
pvalueCutoff = 1,
qvalueCutoff = 1)
kegg_data <- as.data.frame(kegg_result)
dim(kegg_data)
write.csv(kegg_data, "./03.tomato.kegg.result.csv")
pdf("05.kegg.tomato.柱状图.pdf",height = 4, width = 6)
barplot(kegg_result, showCategory= 10,
drop=F,
color="pvalue",
font.size=10)
dev.off()
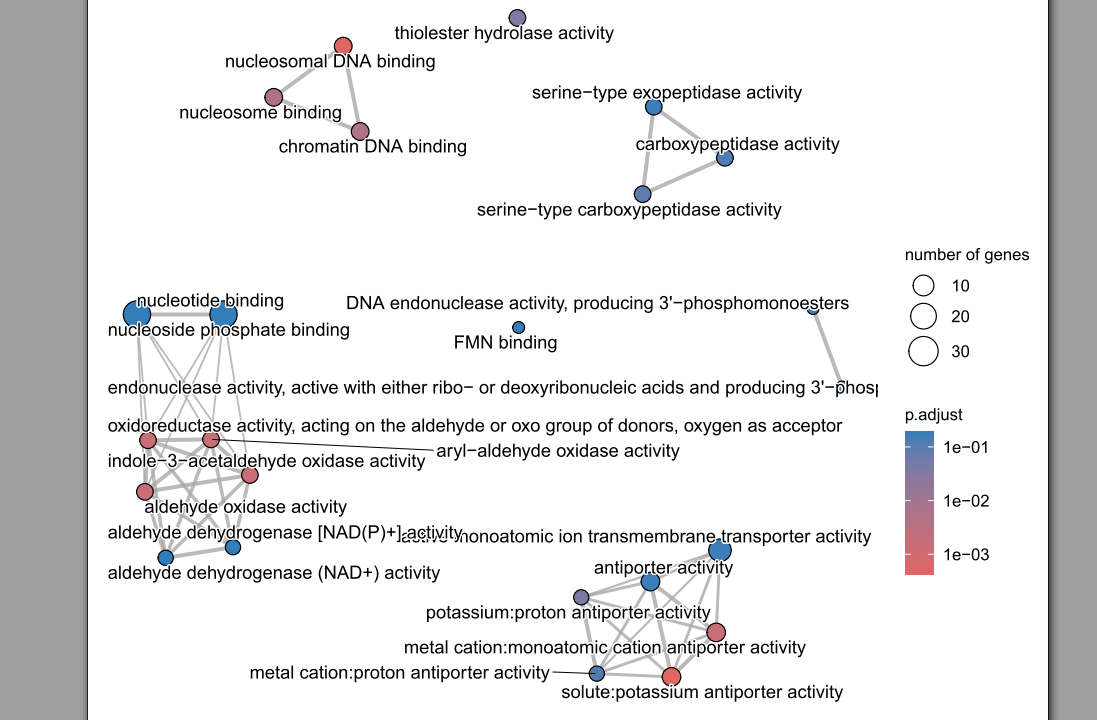
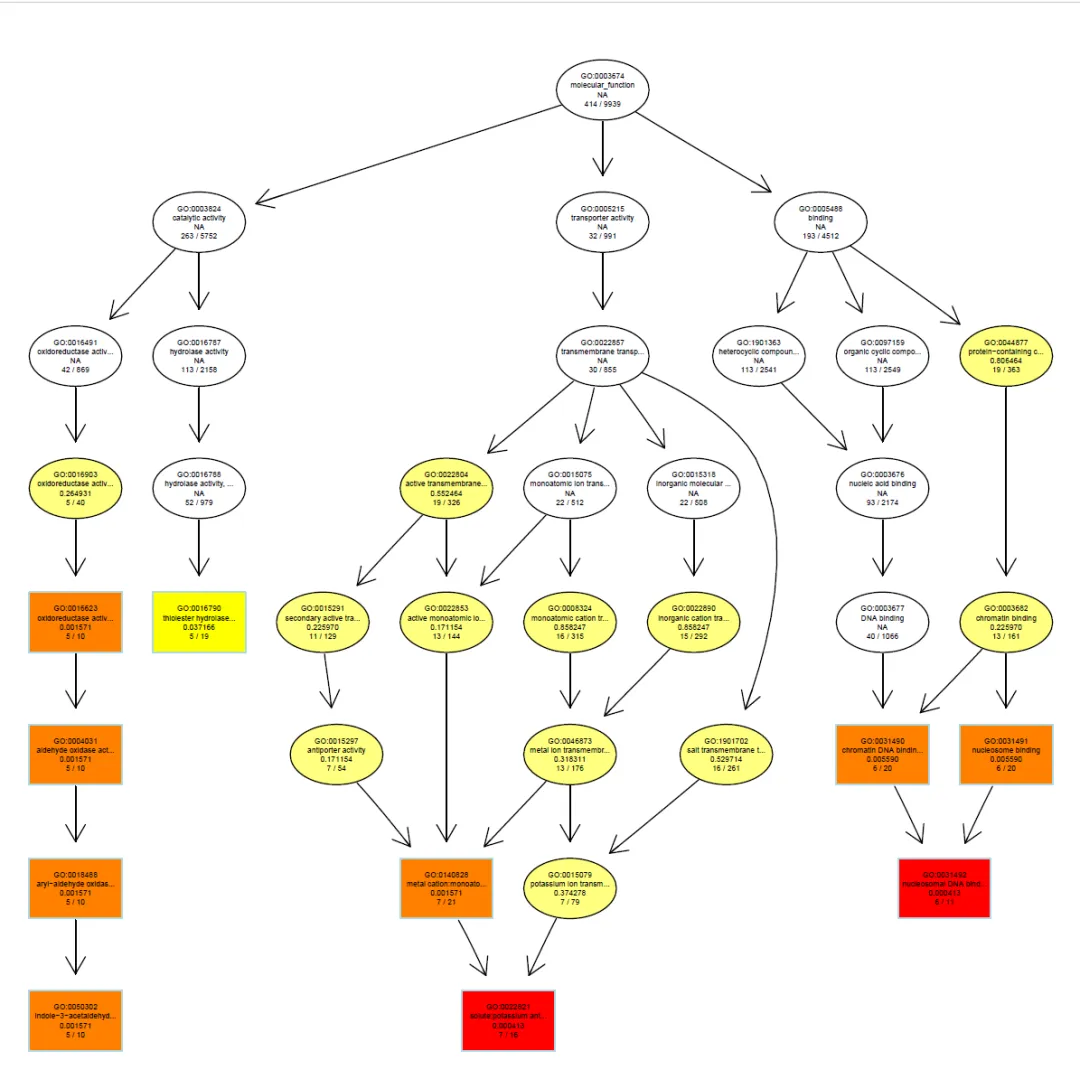
富集网络图
MF_ego <- enrichGO(
gene = gene_list,
keyType = "GID",
OrgDb = org.Slycompersicum.eg.db,
ont = "MF", ##'@GO的种类,BP,CC,MF
pAdjustMethod = "BH",
pvalueCutoff = 0.5##'@结合自己的需求进行调整
)
pdf("./功能富集/05.MF_网络图.pdf", width = 8, height = 8)
plotGOgraph(MF_ego)
dev.off()
MF_ego <- pairwise_termsim(MF_ego)
pdf("./功能富集/06.网络图.pdf",width = 8, height = 8)
emapplot(MF_ego, cex.params = list(category_label = 0.8, line = 0.5)) +
scale_fill_continuous(low = "#e06663", high = "#327eba", name = "p.adjust",
guide = guide_colorbar(reverse = TRUE, order=2), trans='log10')
dev.off()

写在最后
分析就是不停的折腾,不停的捣鼓。小杜在这里记录是自己分析中所学的、用的,以及存在的问题。我们的代码可能在运行时候会出现不同的问题,也会出现,在我这里正常运行,而在你那边程序中却报错的情况。
对于我自己而言,也会出现这样的情况。自己的代码或教程,在前面分析过程中可以运行,但是现在再去使用时也会报错。
好比,这个教程,我昨天的运行时,也各种报错,内心是发狂的,不知道为什么。最后,仅仅只是更改了某个地方的参数,导致在制作过程中缺失变量。
获得本教程 Data and Code,请在后台回复:20240811。
若我们的教程对你有所帮助,请 点赞+收藏+转发,这是对我们最大的支持。
往期部分文章
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
2. 精美图形绘制教程
3. 转录组分析教程
4. 转录组下游分析
小杜的生信筆記 ,主要发表或收录生物信息学教程,以及基于R分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!






 苏公网安备32011502012024号
苏公网安备32011502012024号