导读
本文将介绍为什么要提取最长转录本,以及如何从 fasta和 gff3文件中提取最长转录本。
1. Why
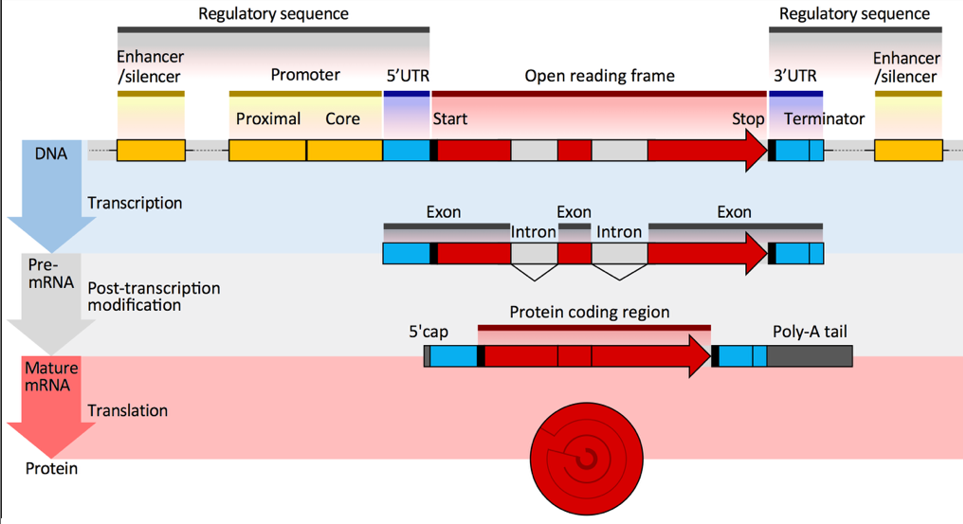
由于可变剪切的存在,通常一个基因可以转录为多个转录本。但是如果将多个转录本同时进行分析,那么分析会因此受到影响。所以,目前的解决办法是,选取一个最具代表性的转录本(最长转录本)来进行分析。
2. 获取方式
- 从序列文件中(
FASTA)提取
- 从基因结构注释信息文件(
GFF)中提取
3. 用法
pip install GetTransTool -i https://pypi.tuna.tsinghua.edu.cn/simple
- 从
GENCODE fasta文件中提取最长转录本
GetLongestTransFromGencode --file example.fa.gz --outfile longest_trans_gencode.fa
# --file 序列文件
# --outfile 输出文件
- 根据
GFF文件(gencode/ensembl/ucsc )提取最长转录本
GetLongestTransFromGTF --database ensembl --gtffile example.gtf.gz --genome example.fa.gz --outfile longest_trans_ensembl.fa
# --database 基因组注释时,选择的数据库
# --gtffile 注释文件
# --genome 基因组序列文件
# --outfile 输出文件
- 从
GENCODE fasta文件中提取最长 CDS
GetCDSLongestFromGencode --file example.fa.gz --outfile longest_cds_trans_gencode.fa
# --file 序列文件
# --outfile 输出文件
- 根据
GFF文件(gencode/ensembl/ucsc )提取最长 CDS
GetCDSLongestFromGTF --database ensembl --gtffile example.gtf.gz --genome example.fa.gz --outfile longest_trans_ensembl.fa
# --database 基因组注释时,选择的数据库
# --gtffile 注释文件
# --genome 基因组序列文件
# --outfile 输出文件

4. code
代码过长,下面只展示部分。

| 







 苏公网安备32011502012024号
苏公网安备32011502012024号