R中单细胞RNA-seq数据分析教程 (2)
## 引言
本系列开启R中单细胞RNA-seq数据分析[教程](https://github.com/quadbio/scRNAseq_analysis_vignette "Source"),持续更新,欢迎关注,转发!
## 导入Seurat包
首先,请确认你的R软件已经安装了Seurat这个包。
```R
library(Seurat)
```
这会将您已安装的Seurat包载入您当前的R环境中。通常不会出现错误,但可能会有一些详细信息显示。如果提示您该包无法找到,请先进行Seurat的安装。
```R
install.packages("Seurat")
library(Seurat)
```
## 1. 创建Seurat对象
Seurat 引入了一种新的数据类型,称为“ Seurat ”。这种数据类型使得 Seurat 能够记录整个分析过程中的所有步骤和结果。因此,首要步骤是导入数据并构建一个 Seurat 对象。对于通过 10x Genomics 平台产生的数据,Seurat 提供了一种便捷的处理方式。
```R
counts <- Read10X(data.dir = "data/DS1/")
seurat <- CreateSeuratObject(counts, project="DS1")
```
`Read10X` 函数的功能是导入数据矩阵,并将其行名和列名分别按照基因名称和细胞条码进行重新命名。当然,如果数据不是通过 10x 平台生成的,人们也可以选择手动进行这些操作。
```R
library(Matrix)
counts <- readMM("data/DS1/matrix.mtx.gz")
barcodes <- read.table("data/DS1/barcodes.tsv.gz", stringsAsFactors=F)[,1]
features <- read.csv("data/DS1/features.tsv.gz", stringsAsFactors=F, sep="\t", header=F)
rownames(counts) <- make.unique(features[,2])
colnames(counts) <- barcodes
seurat <- CreateSeuratObject(counts, project="DS1")
```
如果您浏览(https://satijalab.org/seurat/v3.1/pbmc3k_tutorial.html),您会发现在 `CreateSeuratObj`函数中增加了一些额外的参数,比如 `min.cells`和 `min.features`。当设置这些参数后,会对数据进行初步筛选,从一开始就排除那些在太少细胞中被检测到的基因,以及那些检测到基因数量过少的细胞。这种做法是可行的,但建议保留所有基因(即保持默认设置或将 `min.cells`设为0)。
## 2. 质量控制
在创建Seurat对象之后,接下来的步骤是对数据进行质量控制。最常见的质量控制包括筛选掉:
1. 检测到的基因数量过少的细胞。这些通常意味着细胞的测序深度不足以进行可靠的特征描述。
2. 检测到的基因数量过多的细胞。这些可能代表双细胞或多细胞(即同一液滴中的两个或多个细胞,因此具有相同的细胞条码)。
3. 线粒体转录本百分比过高的细胞。由于大多数单细胞RNA测序实验使用寡T来捕获mRNA,线粒体转录本应该因为缺少poly-A尾而较少,但不可避免地会捕获一些线粒体转录本。同时,也有证据显示一些线粒体转录本中存在稳定的poly-A尾,但它们是降解的标志。总的来说,线粒体转录本百分比高的细胞可能代表处于压力状态的细胞(例如缺氧),这些细胞产生大量的线粒体,或者产生异常高数量的截短线粒体转录本。
虽然Seurat在创建Seurat对象时会自动统计检测到的基因数量(其中nFeature_RNA代表检测到的基因/特征数量;nCount_RNA代表检测到的转录本数量),但线粒体转录本的百分比需要手动计算。不过,Seurat提供了一个便捷的解决方案来处理这个问题。
```R
seurat[["percent.mt"]] <- PercentageFeatureSet(seurat, pattern = "^MT[-\\.]")
```
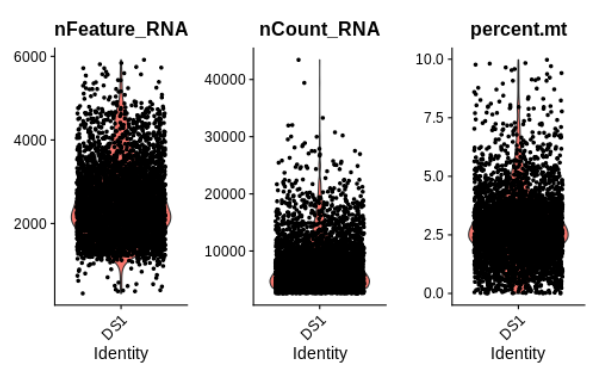
请留意,筛选标准并非一成不变,因为这些指标的正常值范围可能因实验而异,这取决于样本来源、使用的试剂以及测序深度。这里建议**仅排除异常值细胞**,也就是那些在某些质量控制指标上明显偏离大多数细胞的**少数细胞**。为了做到这一点,首先需要了解这些值在数据中的分布情况。可以通过为每个指标绘制小提琴图来观察它们的分布。
```R
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
```
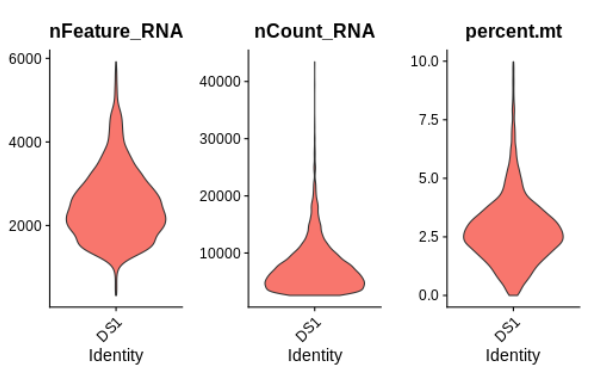
```R
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3, pt.size=0)
```
正如所料,细胞中检测到的基因数量与转录本数量之间存在较强的相关性,但线粒体转录本的百分比却并非如此。
```R
library(patchwork)
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
```

鉴于基因数量与转录本数量之间存在相关性,只需针对其中一个指标设定一个阈值,并结合线粒体转录本百分比的上限来进行质量控制。以这个数据集为例,将检测到的基因数量设定在500到5000之间,线粒体转录本百分比低于5%是一个合理的选择,但根据具体情况调整阈值也是完全可以的。
```R
seurat <- subset(seurat, subset = nFeature_RNA > 500 & nFeature_RNA < 5000 & percent.mt < 5)
```
需要指出的是,有时候可能需要执行更严格的质量控制。其中一个可能的问题是细胞双重捕获(doublets)。由于不同细胞捕获的RNA量差异很大,双重捕获的细胞并不总是表现出更高的基因或转录本检测数量。目前有几个工具可以预测一个“细胞”是否为单个细胞,或者实际上是双重捕获/多重捕获。例如,(https://github.com/chris-mcginnis-ucsf/DoubletFinder) 通过在数据中随机平均细胞构建人工双重捕获,然后测试每个细胞是否与人工双重捕获更相似来预测双重捕获。这有助于判断一个细胞是否有可能是双重捕获。同样,线粒体转录本的百分比也许不足以筛选出处于压力或不健康的细胞。有时,可能需要进行额外的筛选,比如基于[机器学习的预测](https://www.nature.com/articles/s41586-019-1654-9#Sec2)。
## 3. 数据标准化
与批量RNA-seq类似,由于不同细胞捕获的RNA量存在差异,不能直接比较不同细胞中每个基因的捕获转录本数量。因此,需要进行标准化处理,以使不同细胞间的基因表达水平可以相互比较。单细胞RNA-seq数据分析中最常用的标准化方法与TPM(每百万读数的转录本数)概念类似 - 即对每个细胞的特征表达量进行标准化,然后乘以一个缩放因子(默认为10000)。最后,将得到的表情水平进行对数转换,以便表达值更符合正态分布。值得一提的是,在进行对数转换之前,每个值都会加上一个伪计数,这样即使在某个细胞中未检测到转录本的基因,在对数转换后仍然可以显示为零值。
```R
seurat <- NormalizeData(seurat)
```
通常,`NormalizeData`函数中有几个参数可以设置,但大多数时候使用默认设置就足够了。
## 4. 特征选择
与批量RNA-seq相比,单细胞RNA-seq的最大优势在于其能够探究样本的细胞异质性,即通过识别具有独特分子特征的细胞群体。然而,并非所有基因在识别不同细胞群体时都具有同等的信息量和贡献。例如,表达水平低的基因以及在所有细胞中表达水平相似的基因,它们提供的信息量较少,可能会模糊不同细胞群体之间的差异。因此,在深入分析scRNA-seq数据之前,进行恰当的特征选择是非常必要的。
在Seurat或者更广泛地说,在单细胞RNA-seq数据分析中,这一步通常涉及到识别表达水平在细胞间变化最大的高变异性特征/基因。
```R
seurat <- FindVariableFeatures(seurat, nfeatures = 3000)
```
通常,Seurat 会计算每个基因在不同细胞中的标准化方差,并挑选出变异性最大的2000个基因作为高变异特征。你可以通过设置 `nfeatures`参数(例如,这里选择了前3000个基因)来轻松调整高变异特征的数量。
关于应该使用多少高变异特征,并没有一个固定的标准。有时需要尝试几次,以确定哪个数量能够提供最清晰和最容易解释的结果。大多数情况下,选择2000到5000之间的数值是合适的,而且使用不同的数值对结果的影响不会太大。
此外,你可以选择在变量特征图中展示这些结果,但这并不是必需的步骤。
```R
top_features <- head(VariableFeatures(seurat), 20)
plot1 <- VariableFeaturePlot(seurat)
plot2 <- LabelPoints(plot = plot1, points = top_features, repel = TRUE)
plot1 + plot2
```

页:
[1]