联合 RNA 和 ATAC 分析:SNARE-seq
## 引言[本文](https://stuartlab.org/signac/articles/snareseq "Source")将带您分析一个单细胞联合检测数据集,该数据集能够同时测量细胞内的基因表达水平和DNA的可及性。
这项数据集由Chen、Lake和Zhang在2019年发表,采用了一种名为SNARE-seq的技术。由于该数据集并未公开,我们已将原始数据重新映射至mm10基因组。您可以通过以下链接下载:
1. 片段文件:https://signac-objects.s3.amazonaws.com/snareseq/fragments.sort.bed.gz
2. 片段文件的索引文件:https://signac-objects.s3.amazonaws.com/snareseq/fragments.sort.bed.gz.tbi
3. 用于从原始数据生成片段文件的代码:https://github.com/timoast/SNARE-seq
## 数据加载
首先构建了一个Seurat对象,它包含了两种不同的检测类型:一种是基因表达数据,另一种是DNA的可及性数据。
在加载计数数据时,我们利用Seurat提供的Read10X()功能。使用这个功能之前,需要将barcodes.tsv.gz、matrix.mtx.gz和features.tsv.gz这些文件整理到一个单独的文件夹中。
```R
library(Signac)
library(Seurat)
library(ggplot2)
library(EnsDb.Mmusculus.v79)
# load processed data matrices for each assay
rna <- Read10X("../vignette_data/snare-seq/GSE126074_AdBrainCortex_rna/", gene.column = 1)
atac <- Read10X("../vignette_data/snare-seq/GSE126074_AdBrainCortex_atac/", gene.column = 1)
fragments <- "../vignette_data/snare-seq/fragments.sort.bed.gz"
# create a Seurat object and add the assays
snare <- CreateSeuratObject(counts = rna)
snare[['ATAC']] <- CreateChromatinAssay(
counts = atac,
sep = c(":", "-"),
genome = "mm10",
fragments = fragments
)
# extract gene annotations from EnsDb
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Mmusculus.v79)
# change to UCSC style since the data was mapped to mm10
seqlevels(annotations) <- paste0('chr', seqlevels(annotations))
genome(annotations) <- "mm10"
# add the gene information to the object
Annotation(snare[["ATAC"]]) <- annotations
```
## 数据质控
```R
DefaultAssay(snare) <- "ATAC"
snare <- TSSEnrichment(snare)
snare <- NucleosomeSignal(snare)
snare$blacklist_fraction <- FractionCountsInRegion(
object = snare,
assay = 'ATAC',
regions = blacklist_mm10
)
Idents(snare) <- "all"# group all cells together, rather than by replicate
VlnPlot(
snare,
features = c("nCount_RNA", "nCount_ATAC", "TSS.enrichment",
"nucleosome_signal", "blacklist_fraction"),
pt.size = 0.1,
ncol = 5
)
```

```R
snare <- subset(
x = snare,
subset = blacklist_fraction < 0.03 &
TSS.enrichment < 20 &
nCount_RNA > 800 &
nCount_ATAC > 500
)
snare
## An object of class Seurat
## 277704 features across 8055 samples within 2 assays
## Active assay: ATAC (244544 features, 0 variable features)
##2 layers present: counts, data
##1 other assay present: RNA
```
## 基因表达数据处理
- 使用 Seurat 处理基因表达数据
```R
DefaultAssay(snare) <- "RNA"
snare <- FindVariableFeatures(snare, nfeatures = 3000)
snare <- NormalizeData(snare)
snare <- ScaleData(snare)
snare <- RunPCA(snare, npcs = 30)
snare <- RunUMAP(snare, dims = 1:30, reduction.name = "umap.rna")
snare <- FindNeighbors(snare, dims = 1:30)
snare <- FindClusters(snare, resolution = 0.5, algorithm = 3)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 8055
## Number of edges: 324240
##
## Running smart local moving algorithm...
## Maximum modularity in 10 random starts: 0.8900
## Number of communities: 14
## Elapsed time: 4 seconds
p1 <- DimPlot(snare, label = TRUE) + NoLegend() + ggtitle("RNA UMAP")
```
## DNA可及性数据处理
- 使用 Signac 处理 DNA 可及性数据
```R
DefaultAssay(snare) <- 'ATAC'
snare <- FindTopFeatures(snare, min.cutoff = 10)
snare <- RunTFIDF(snare)
snare <- RunSVD(snare)
snare <- RunUMAP(snare, reduction = 'lsi', dims = 2:30, reduction.name = 'umap.atac')
p2 <- DimPlot(snare, reduction = 'umap.atac', label = TRUE) + NoLegend() + ggtitle("ATAC UMAP")
p1 + p2
```
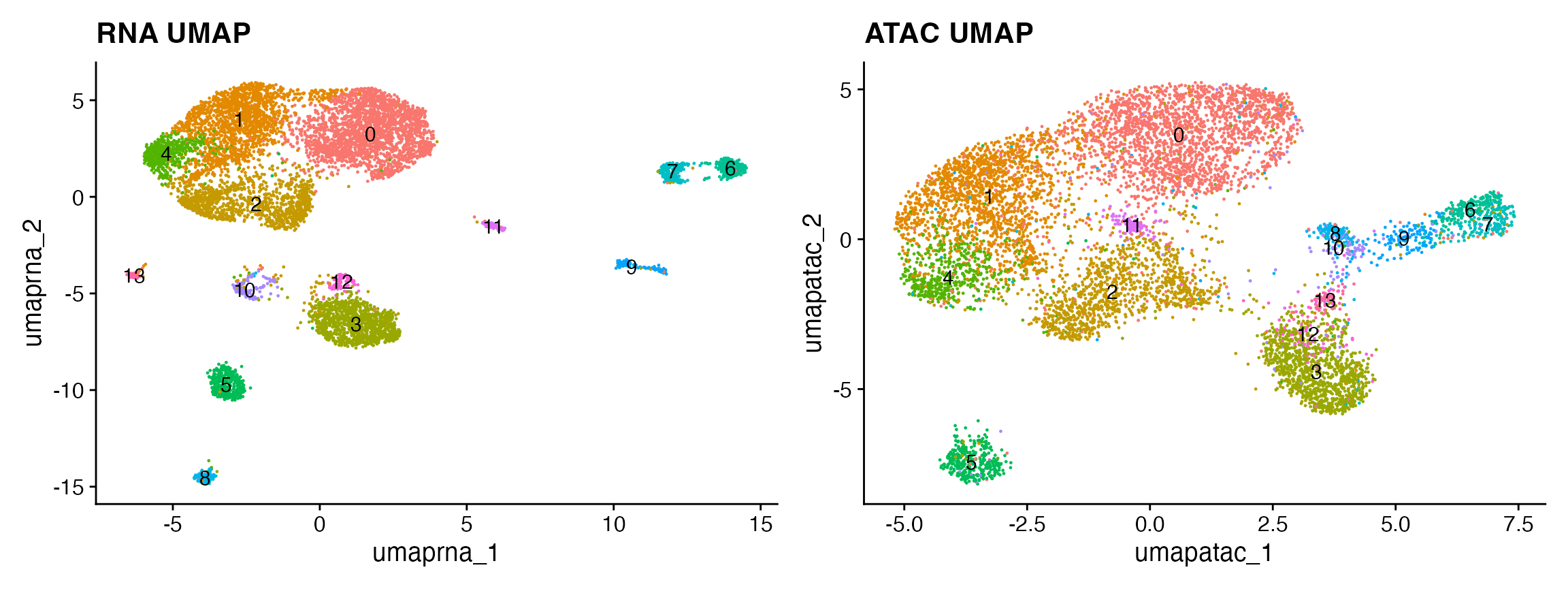
## 与 scRNA-seq 整合
接下来,可以通过成人小鼠大脑的单细胞RNA测序(scRNA-seq)数据集的标签,来对当前数据集中的细胞类型进行分类标注。
```R
# label transfer from Allen brain
allen <- readRDS("../vignette_data/allen_brain.rds")
allen <- UpdateSeuratObject(allen)
# use the RNA assay in the SNARE-seq data for integration with scRNA-seq
DefaultAssay(snare) <- 'RNA'
transfer.anchors <- FindTransferAnchors(
reference = allen,
query = snare,
dims = 1:30,
reduction = 'cca'
)
predicted.labels <- TransferData(
anchorset = transfer.anchors,
refdata = allen$subclass,
weight.reduction = snare[['pca']],
dims = 1:30
)
snare <- AddMetaData(object = snare, metadata = predicted.labels)
# label clusters based on predicted ID
new.cluster.ids <- c(
"L2/3 IT",
"L4",
"L6 IT",
"L5 CT",
"L4",
"L5 PT",
"Pvalb",
"Sst",
"Astro",
"Oligo",
"Vip/Lamp5",
"L6 IT.2",
"L6b",
"NP"
)
names(x = new.cluster.ids) <- levels(x = snare)
snare <- RenameIdents(object = snare, new.cluster.ids)
snare$celltype <- Idents(snare)
DimPlot(snare, group.by = 'celltype', label = TRUE, reduction = 'umap.rna')
```
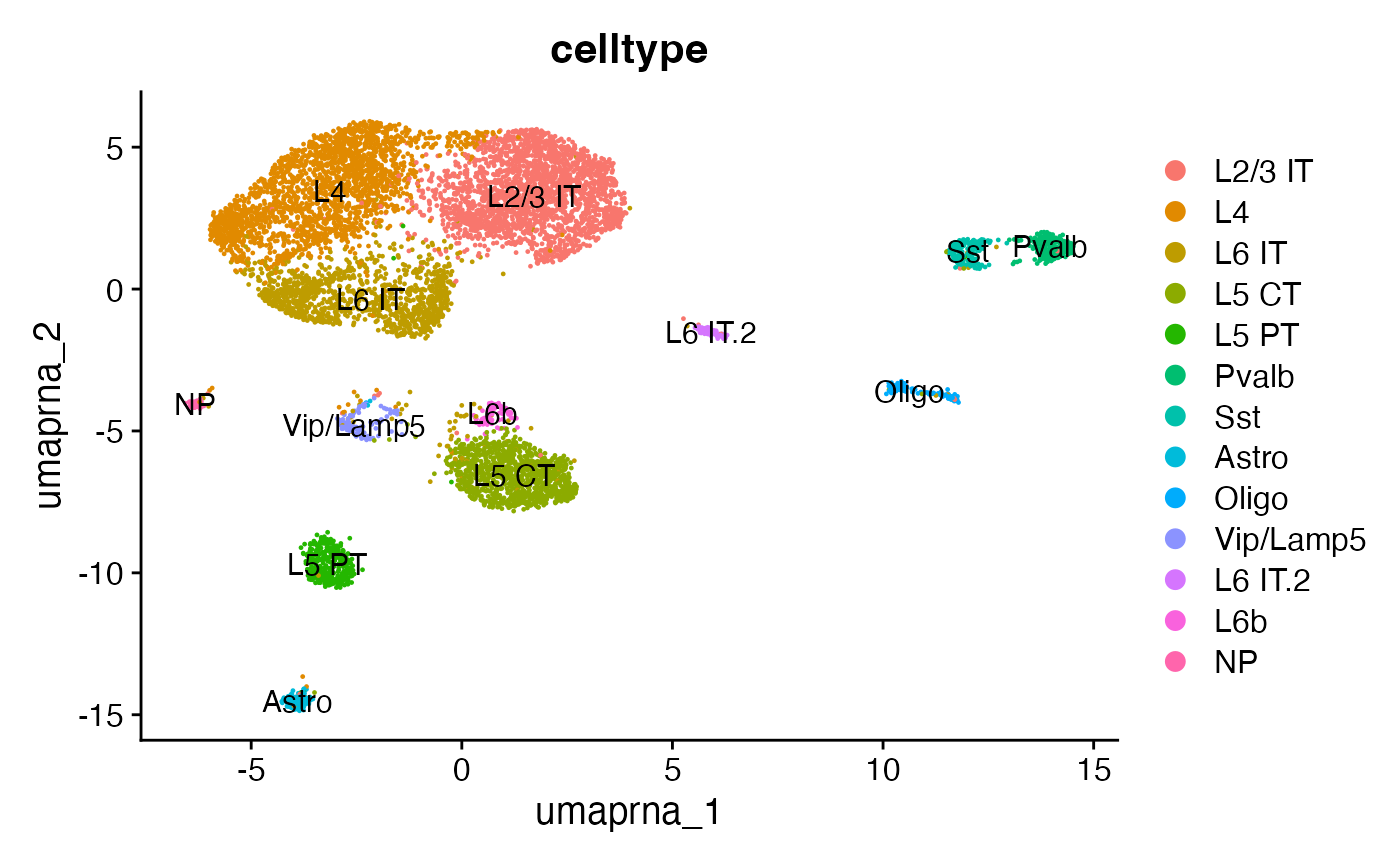
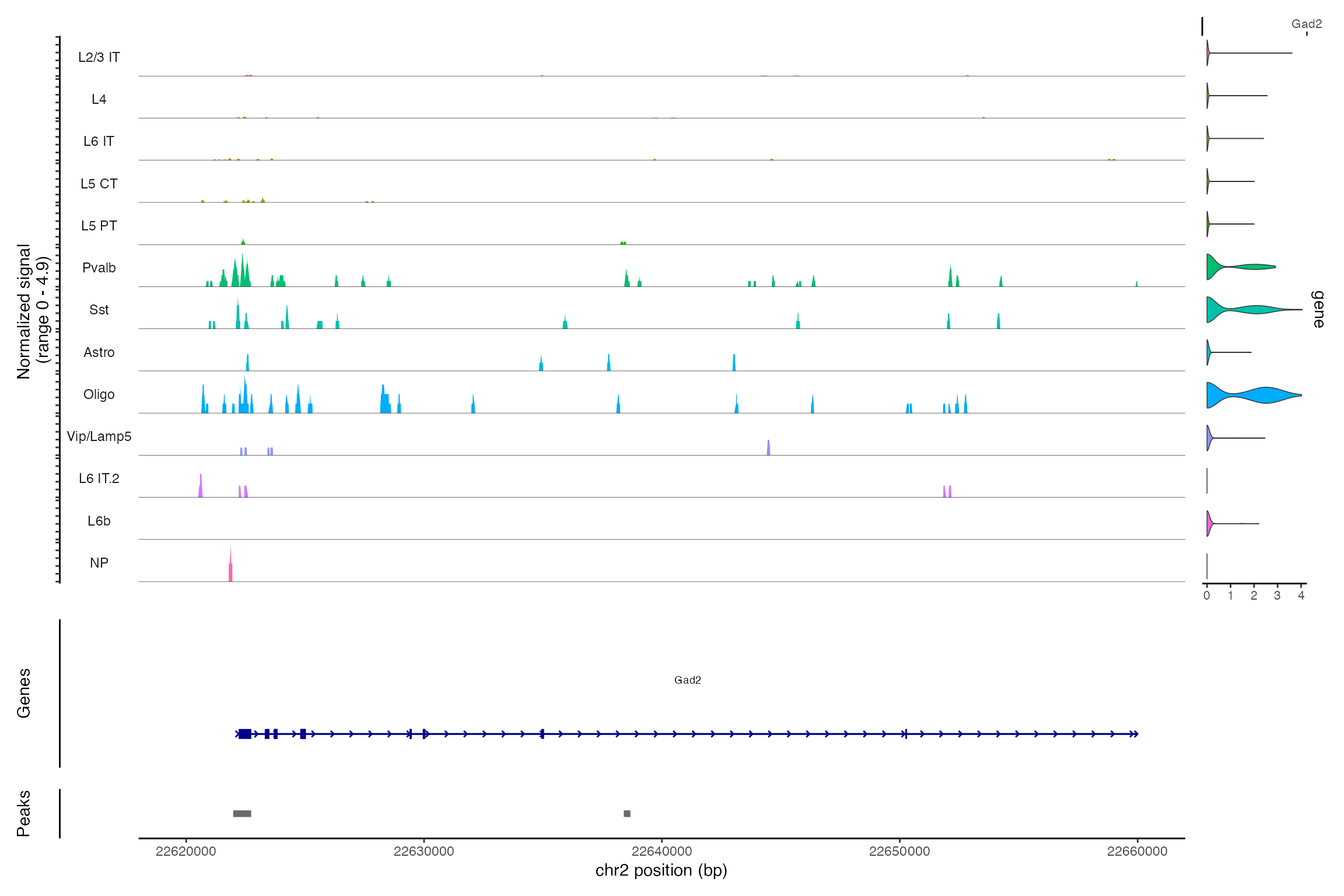
## 同时展示基因表达和DNA开放性
利用CoveragePlot()功能,我们可以同时观察基因表达和DNA可及性数据。这种方式便于对不同细胞类型在特定区域内的DNA开放性进行比较,并且能够将不同基因的表达情况叠加显示,以便于分析。
```R
DefaultAssay(snare) <- "ATAC"
CoveragePlot(snare, region = "chr2-22620000-22660000", features = "Gad2")
```
页:
[1]