空间单细胞|Slide-seq分析、可视化与整合(2)
## 数据集在本文中,我们将对利用Slide-seq v2技术获得的小鼠海马区数据集进行深入分析。
为了方便数据获取,您可以利用我们的SeuratData包,具体操作示例如下。一旦安装了该数据集,您只需输入 `?ssHippo`,即可查看构建Seurat对象时所使用的命令列表。
```R
InstallData("ssHippo")
slide.seq <- LoadData("ssHippo")
```
## 空间变化特征的识别
正如Visium教程中所述,我们可以通过两种主要方法来识别具有空间变异性的特征:一种是在预先标记的解剖区域之间进行差异表达分析,另一种是采用统计方法来评估特征与空间位置的相关性。
在本例中,我们展示了后一种方法,即通过FindSpatiallyVariableFeatures()函数实现的Moran's I统计量,只需设置方法为 `'moransi'`即可。Moran's I能够计算整体的空间自相关性,并提供一个类似于相关系数的统计值,用以衡量特征与空间位置的相关程度。这允许我们根据特征表达的空间变异性进行排序。为了快速估算这一统计值,我们采用了一种基本的分箱策略,该策略会在Slide-seq puck上绘制一个矩形网格,并计算每个网格内特征和位置的平均值。x轴和y轴上的分箱数量可以通过x.cuts和y.cuts参数分别控制。另外,虽然不是强制要求,但安装可选的Rfast2包(通过运行 `install.packages('Rfast2')`)可以显著提高计算效率,从而减少运行时间。
```R
DefaultAssay(slide.seq) <- "SCT"
slide.seq <- FindSpatiallyVariableFeatures(slide.seq, assay = "SCT", slot = "scale.data", features = VariableFeatures(slide.seq),
selection.method = "moransi", x.cuts = 100, y.cuts = 100)
```
现在我们可视化 Moran’s I 识别的前 6 个特征的表达。
```R
SpatialFeaturePlot(slide.seq, features = head(SpatiallyVariableFeatures(slide.seq, selection.method = "moransi"),
6), ncol = 3, alpha = c(0.1, 1), max.cutoff = "q95")
```

## 使用 RCTD 进行空间反卷积
FindTransferAnchors功能可以用于整合来自空间转录组数据集的单个斑点数据,而Seurat v5版本还增加了对Robust Cell Type Decomposition(鲁棒细胞类型分解)的支持。这是一种计算技术,当提供单细胞RNA测序(scRNA-seq)参考数据时,可以用来从空间数据集中反推斑点级别的数据。RCTD已被证实能够精确地对来自SLIDE-seq、Visium和10x公司的Xenium in-situ空间平台等多种技术的空间数据进行注释。
要执行RCTD,我们首先需要从GitHub上安装spacexr包,这个包提供了RCTD的实现。
```R
devtools::install_github("dmcable/spacexr", build_vignettes = FALSE)
```
从 Seurat 查询和参考对象中提取计数、聚类和点信息,以构建 RCTD 用于注释的参考和 SpatialRNA 对象。
```R
library(spacexr)
# set up reference
ref <- readRDS("../data/mouse_hippocampus_reference.rds")
ref <- UpdateSeuratObject(ref)
Idents(ref) <- "celltype"
# extract information to pass to the RCTD Reference function
counts <- ref[["RNA"]]$counts
cluster <- as.factor(ref$celltype)
names(cluster) <- colnames(ref)
nUMI <- ref$nCount_RNA
names(nUMI) <- colnames(ref)
reference <- Reference(counts, cluster, nUMI)
# set up query with the RCTD function SpatialRNA
slide.seq <- SeuratData::LoadData("ssHippo")
counts <- slide.seq[["Spatial"]]$counts
coords <- GetTissueCoordinates(slide.seq)
colnames(coords) <- c("x", "y")
coords <- NULL
query <- SpatialRNA(coords, counts, colSums(counts))
```
利用参照数据集和待分析的数据集,我们对数据集进行了细胞类型的标注,并将这些标签整合到了待查询的Seurat对象中。Robust Cell Type Decomposition(RCTD)算法支持并行计算,这意味着可以通过分配更多的处理器核心来提升运算速度。
```R
RCTD <- create.RCTD(query, reference, max_cores = 8)
RCTD <- run.RCTD(RCTD, doublet_mode = "doublet")
slide.seq <- AddMetaData(slide.seq, metadata = RCTD@results$results_df)
```
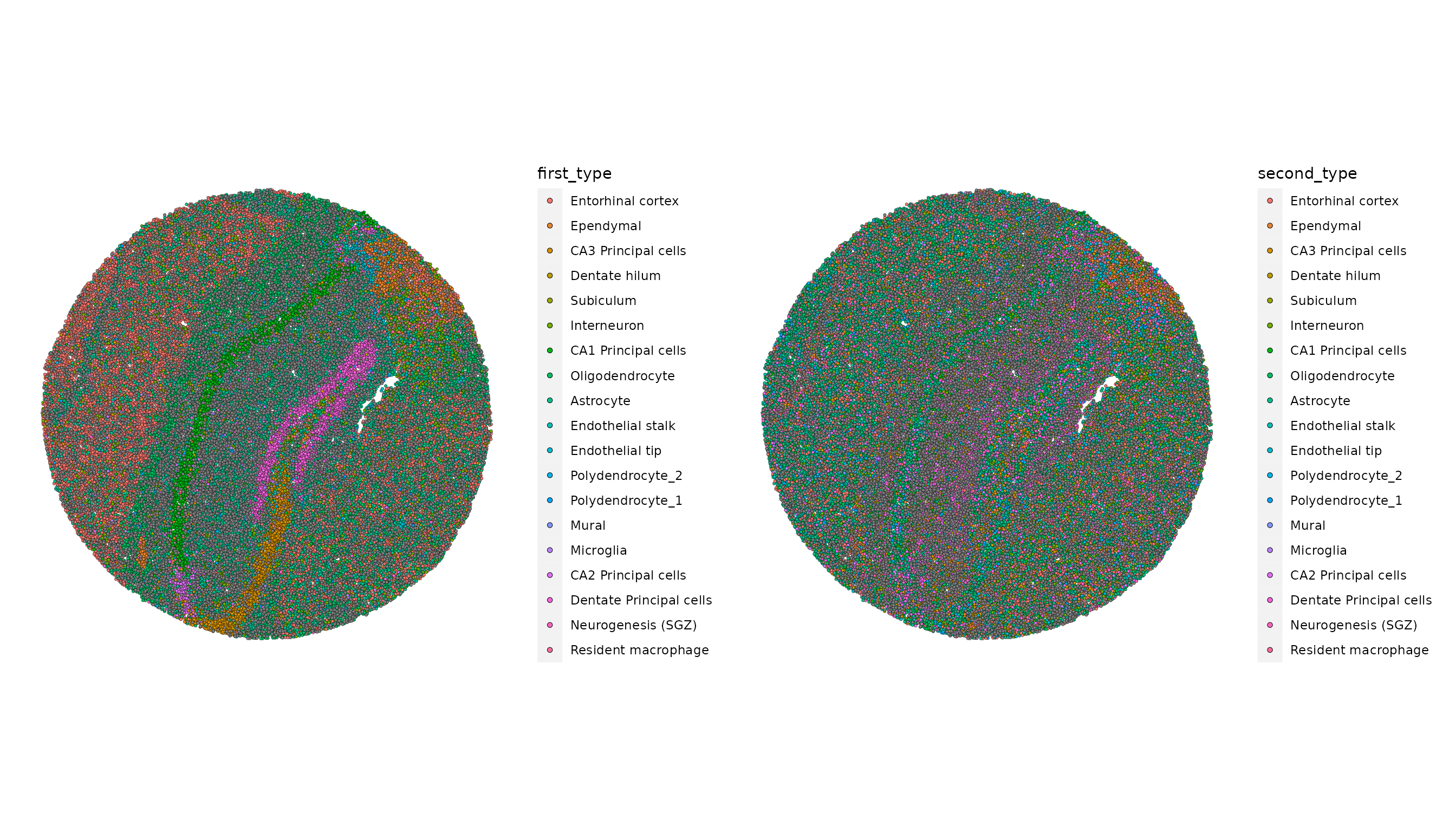
然后,展示RCTD的注释结果。由于我们在双细胞检测模式下执行了RCTD分析,该算法为每个条形码或斑点指定了主要细胞类型(first_type)和次要细胞类型(second_type)。
```R
p1 <- SpatialDimPlot(slide.seq, group.by = "first_type")
p2 <- SpatialDimPlot(slide.seq, group.by = "second_type")
p1 | p2
```
页:
[1]