空间单细胞|10x Visium数据分析、可视化与整合(2)
## 引言[本文](https://satijalab.org/seurat/articles/spatial_vignette#overview "Source")介绍了使用Seurat分析具有空间分辨率的RNA测序数据的方法,重点在于将空间信息与分子数据相结合。将包括以下常见于空间数据分析的任务:
- 数据标准化
- 降维和数据聚类
- 发现空间变异性特征
- 与单细胞RNA测序数据的整合
- 处理多个样本切片
## 降维;聚类;可视化
然后,可以使用与 scRNA-seq 分析相同的工作流程,对 RNA 表达数据进行降维和聚类。
```R
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE)
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
```
然后,可以在 UMAP 空间(使用 DimPlot())中可视化聚类结果,或者使用 SpatialDimPlot() 将聚类结果叠加在图像上。
```R
p1 <- DimPlot(brain, reduction = "umap", label = TRUE)
p2 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
p1 + p2
```
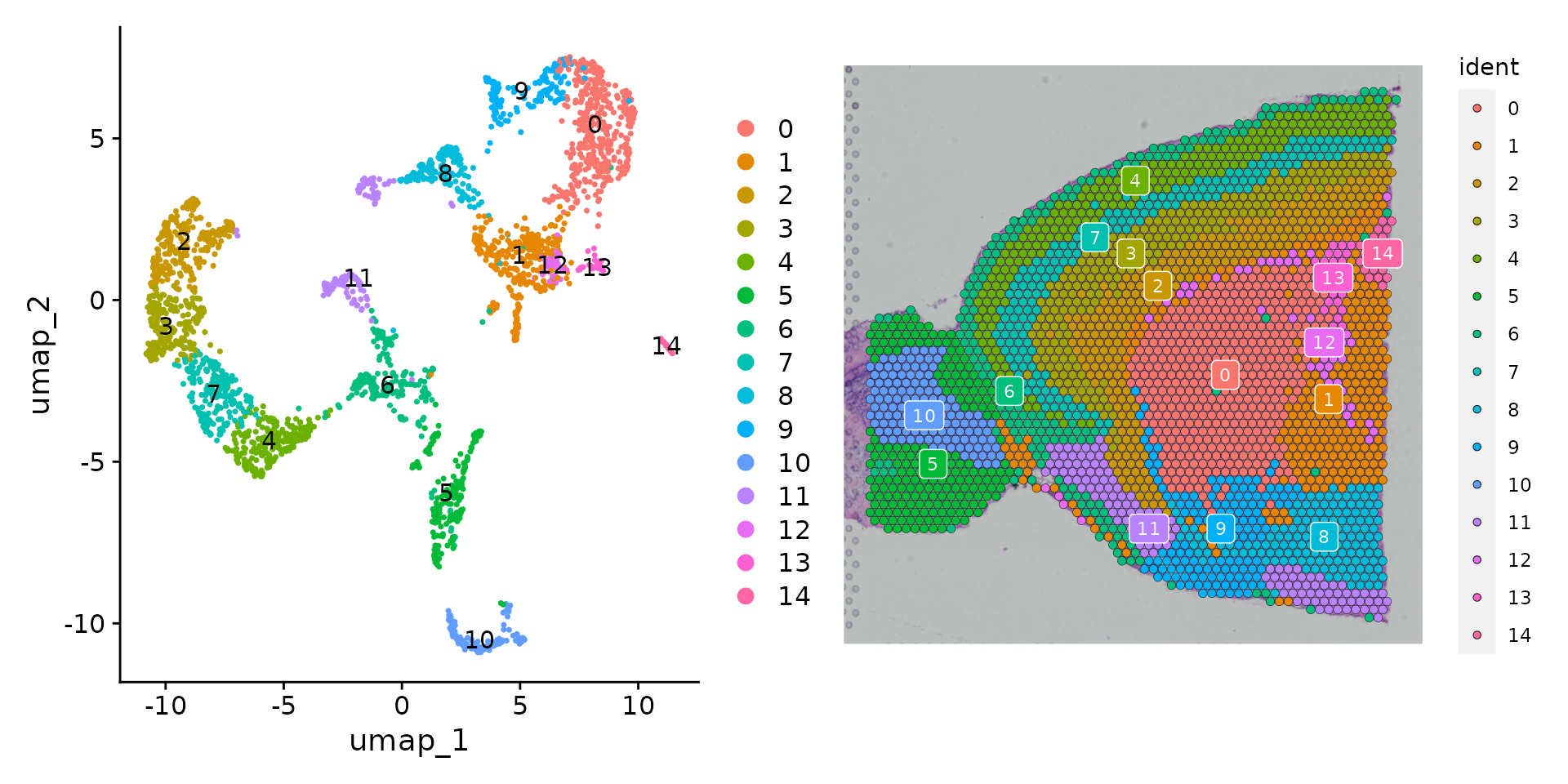
面对众多颜色,要弄清楚每个体素属于哪个簇可能会有些困难。提供了几种方法来应对这一挑战。通过设置标签参数,可以在每个簇的中心位置放置一个彩色框以示区分(参见上方的图表)。
此外,您还可以利用cells.highlight参数在SpatialDimPlot()图中突出显示某些特别关注的细胞。这在识别不同簇的空间分布时非常有用,正如在下面的示例中展示的那样。
```R
SpatialDimPlot(brain, cells.highlight = CellsByIdentities(object = brain, idents = c(2, 1, 4, 3,
5, 8)), facet.highlight = TRUE, ncol = 3)
```
!(https://s2.loli.net/2024/06/24/rkwyaxJMROc4noB.png)
## 空间变化特征的识别
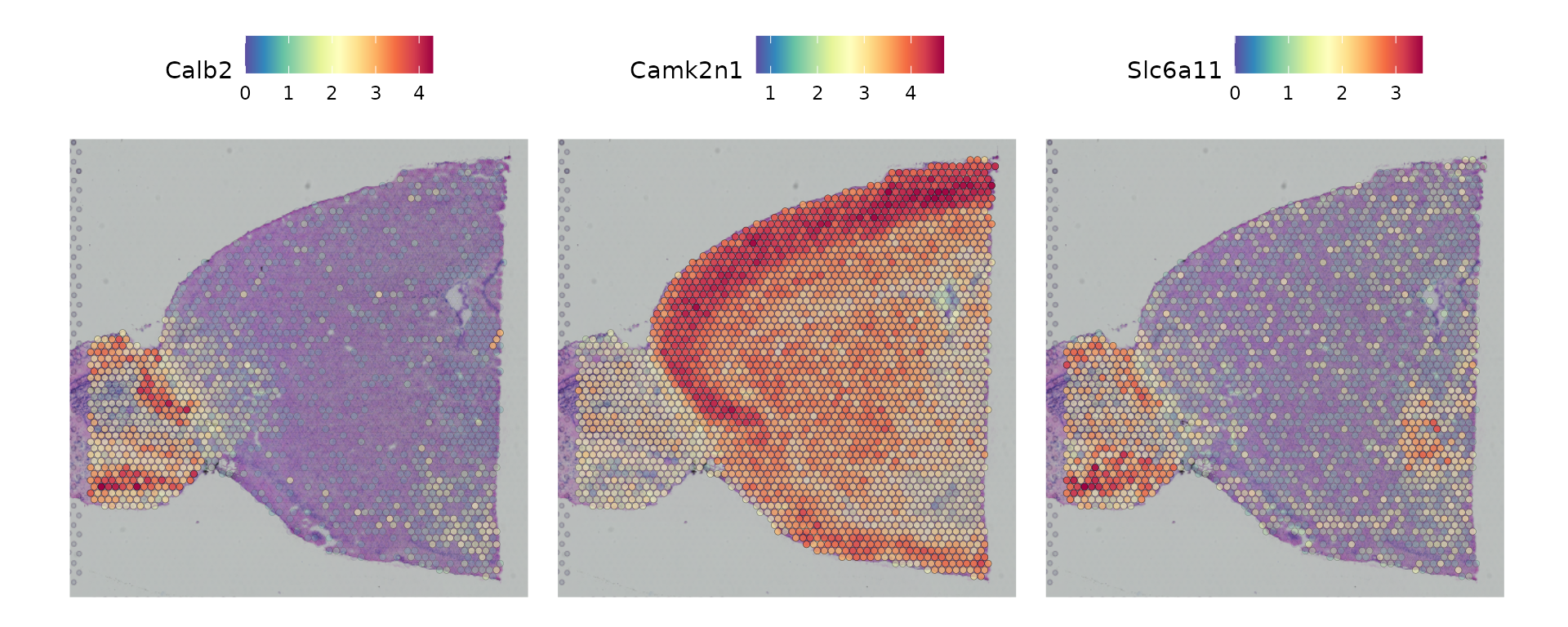
Seurat 提供了两种方法来识别与组织内部空间位置相联系的分子特征。第一种方法是根据组织内预先定义的解剖区域进行差异表达分析,这些区域的确定可以基于无监督聚类的结果或者已有的知识。在当前情况下,这种方法是可行的,因为所讨论的簇具有清晰可见的空间分布特征。
```R
de_markers <- FindMarkers(brain, ident.1 = 5, ident.2 = 6)
SpatialFeaturePlot(object = brain, features = rownames(de_markers), alpha = c(0.1, 1), ncol = 3)
```
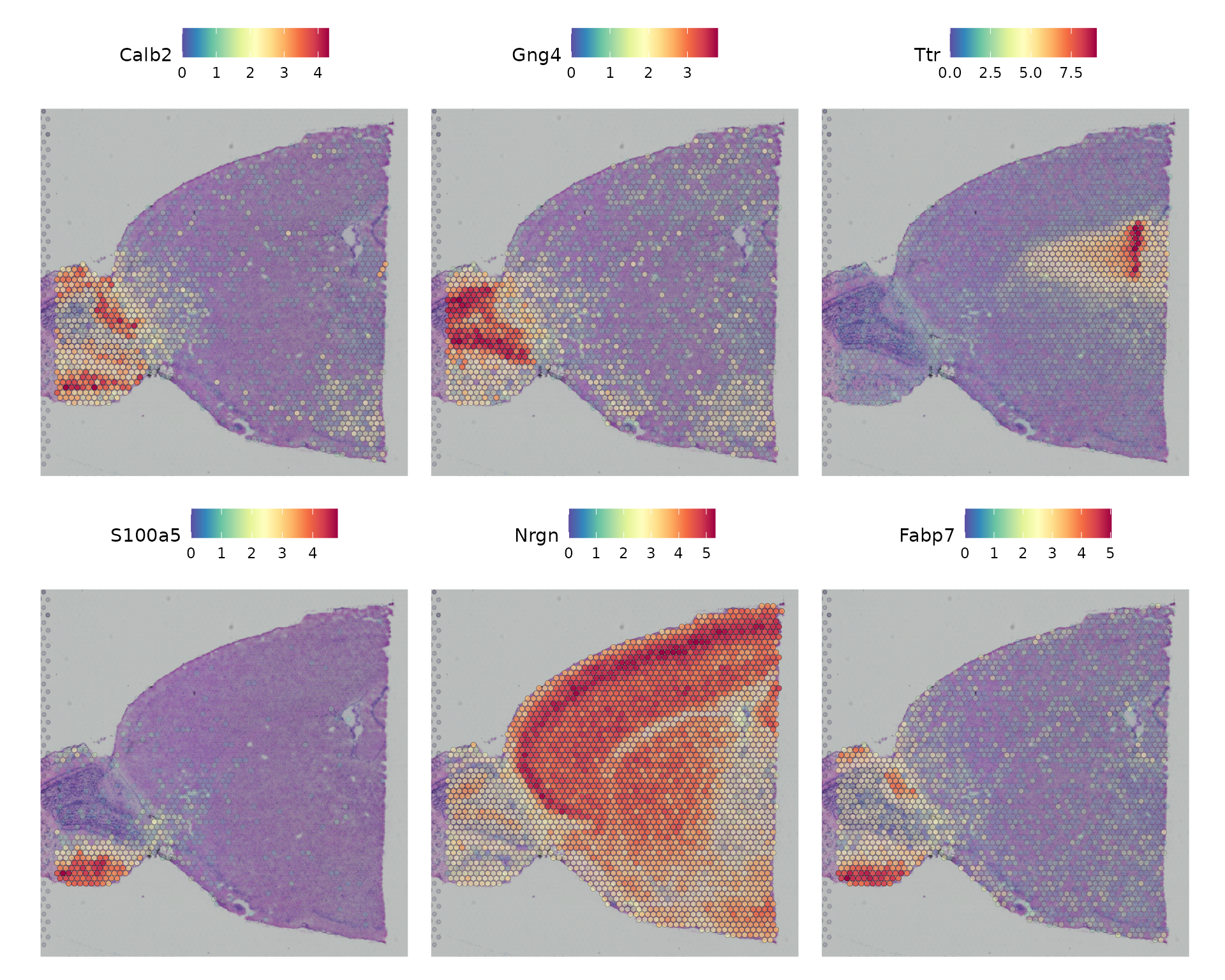
除了基于预标注区域的差异表达分析,Seurat还提供了另一种方法FindSpatiallyVariables(),用于在没有预先标注的情况下寻找具有空间分布模式的分子特征。这种方法的默认实现(method = 'markvariogram'),借鉴了Trendsceek算法的思想,它将空间转录组数据视作标记点过程,并通过计算“变差图”来识别那些其表达水平与空间位置相关的基因。具体来说,这一过程会计算gamma(r)值,用以衡量在特定距离“r”处两个点之间的相关性。默认情况下,在分析中使用距离“r”值为5,并仅针对变量基因(其变异性是独立于空间位置来计算的)进行计算,以提高效率。
```R
brain <- FindSpatiallyVariableFeatures(brain, assay = "SCT", features = VariableFeatures(brain),
selection.method = "moransi")
```
现在可视化通过该度量识别的前 6 个特征的表达。
```R
top.features <- head(SpatiallyVariableFeatures(brain, selection.method = "moransi"), 6)
SpatialFeaturePlot(brain, features = top.features, ncol = 3, alpha = c(0.1, 1))
```
## 提取特定解剖区域
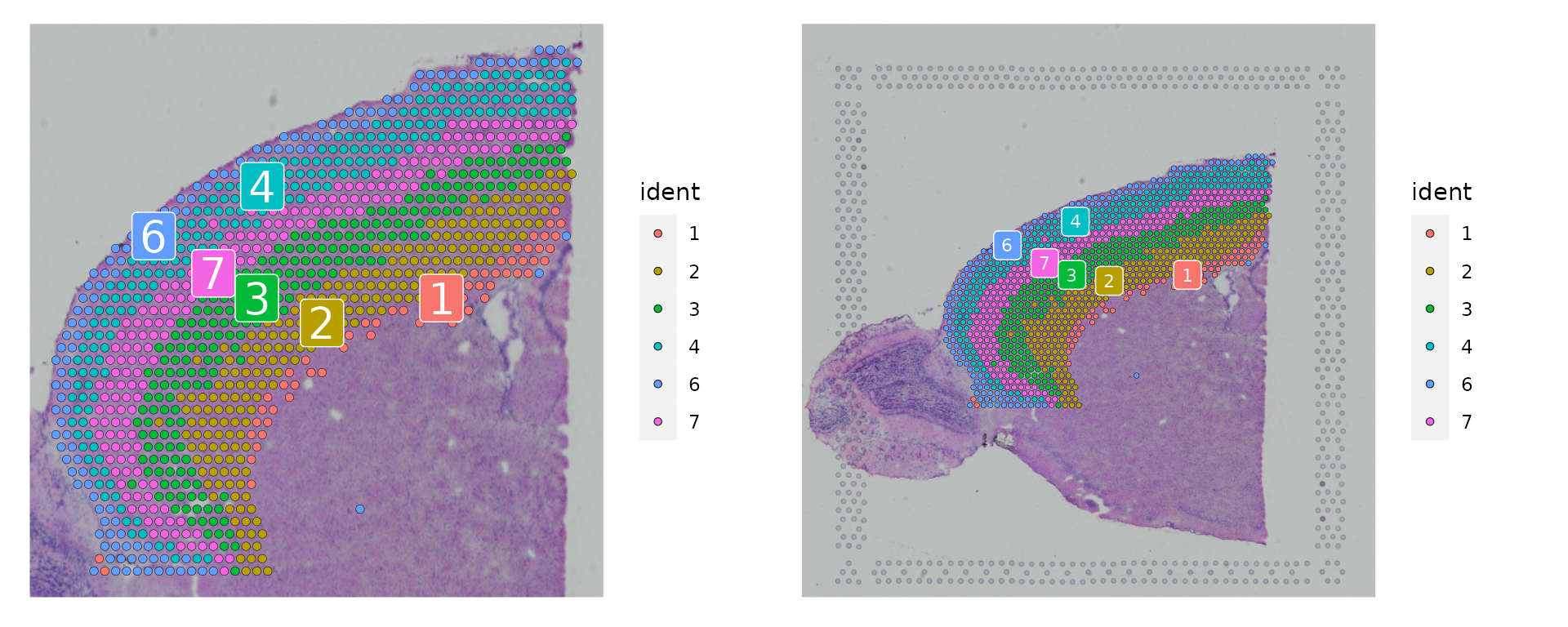
与处理单细胞数据对象相似,您可以选择性地关注数据的特定部分。例如,可以大致选取前额皮层区域进行分析。这种做法也为之后将这些数据与皮层单细胞RNA测序数据集进行整合提供了便利。首先选择一部分细胞簇,然后根据它们确切的空间位置进行细分。完成这一子集操作后,可以选择在完整的图像上或者图像的裁剪部分上展示皮层细胞的分布情况。
```R
cortex <- subset(brain, idents = c(1, 2, 3, 4, 6, 7))
# now remove additional cells, use SpatialDimPlots to visualize what to remove
# SpatialDimPlot(cortex,cells.highlight = WhichCells(cortex, expression = image_imagerow > 400
# | image_imagecol < 150))
cortex <- subset(cortex, anterior1_imagerow > 400 | anterior1_imagecol < 150, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 275 & anterior1_imagecol > 370, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 250 & anterior1_imagecol > 440, invert = TRUE)
p1 <- SpatialDimPlot(cortex, crop = TRUE, label = TRUE)
p2 <- SpatialDimPlot(cortex, crop = FALSE, label = TRUE, pt.size.factor = 1, label.size = 3)
p1 + p2
```
页:
[1]